
1.研究背景
南京理工大学材料科学与工程学院纳米异构中心任吉昌团队撰写题为“A data-driven framework for efficient and physically interpretable prediction of RPA-level adsorption energies”的研究文章,并发表于Materials Today Electronics期刊。该工作提出了一种融合多保真度机器学习与符号回归的数据驱动新框架,在显著降低计算成本的同时,实现了接近随机相位相似(RPA)精度级别的吸附能预测,并给出了具有明确物理含义的解析表达式,为高效催化材料设计提供了新思路。
反应中间体在金属表面的吸附能是决定催化性能的关键参数,然而,目前主流的高精度理论方法——RPA虽然能够将误差降低至约0.1eV,但其计算代价极为昂贵,难以支撑大规模材料筛选。相比之下,常规密度泛函方法计算效率高,但却存在系统性误差。针对这一长期存在的矛盾,研究团队构建了一个多保真高斯过程(MFGP)模型,将大量廉价的低精度DFT数据(如LDA)与少量高精度RPA数据有机结合。结果表明,仅使用9%的RPA数据,模型即可实现R2≈0.96的预测精度,大幅降低了对昂贵高精度计算的依赖。
除提升预测效率外,该工作更进一步解决了机器学习在催化领域普遍存在的“可解释性”不足的问题。研究团队结合特征重要性分析(SHAP)与符号回归(SISSO),从多种原子描述符中筛选出最关键的物理变量,并最终推导出一个简洁的解析公式,E_ads^SISSO=SE×(k_1 VE+k_2 (VE/Rs))+b,该公式清晰揭示了吸附强度与金属价电子数、Wigner-Seitz半径及表面能之间的内在联系,为传统d带理论补充了新的定量补充,也为理解不同层级之间的系统性偏差提供了物理图像。
研究还系统分析了不同交换关联泛函与RPA之间的映射关系,揭示了低阶泛函在CO、NO等吸附体系中误差显著放大的规律,为后续高精度数据的针对性补充提供了指导。该框架不仅适用于传统过渡金属表面,也有望推广至更复杂的合金表面催化体系,为构建快速、低成本且具备物理可解释性的材料筛选平台奠定基础。
2.图文速递
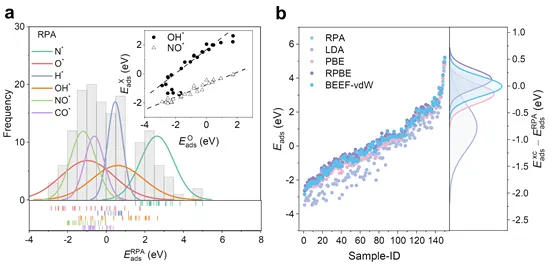
图1.(a)RPA泛函下小分子吸附能的分布,以及O*和OH*/ NO*之间吸附能的线性标度关系。(b)不同泛函之间小分子吸附能的差异。
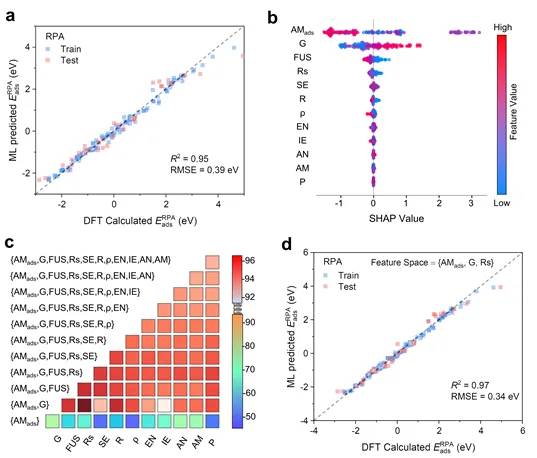
图2.(a)基于RPA泛函吸附能训练的随机森林模型的预测性能。(b)基于训练的随机森林模型的SHAP分析。(c)基于随机森林模型的特征组合筛选过程。(d)使用{AM_ads,G,Rs}特征组合的随机森林模型的预测性能。
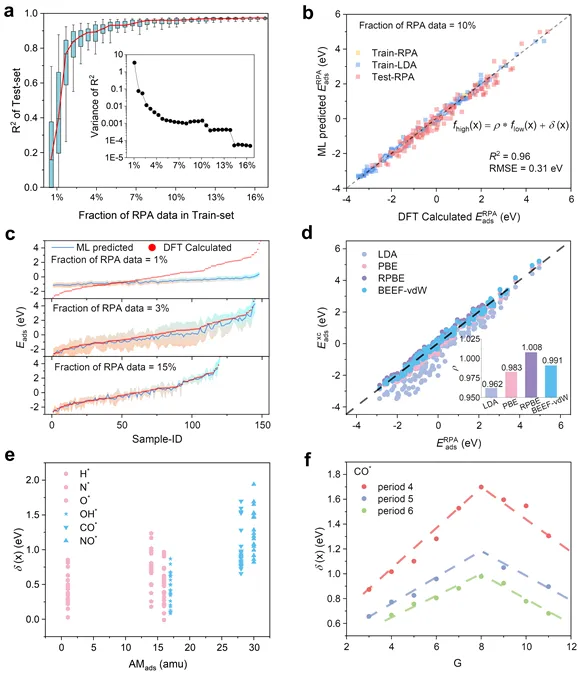
图3.(a)MFGP模型基于不同数量的高保真RPA数据下的预测性能。(b)MFGP模型对高保证数据和低保真数据的综合预测性能。(c)MFGP模型在不同高保真数据数量下的预测置信区间。(d)基于MFGP模型的不同泛函和RPA泛函之间吸附能的线性标度关系。(e)MFGP模型训练过程中不同吸附质的差异项分布。(f)MFGP模型训练过程中不同表面的差异项分布。
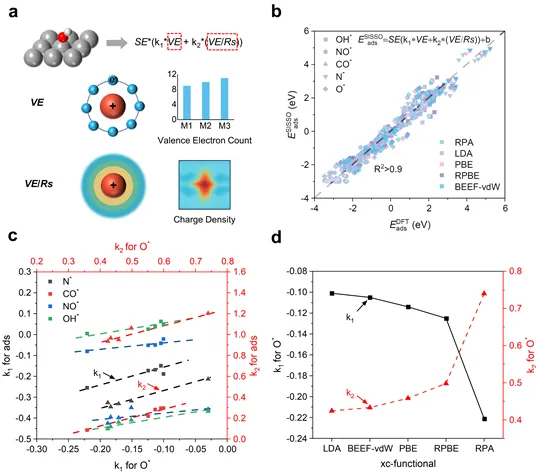
图4.(a)SISSO吸附能公式描述符VE和VE/Rs。(b)不同泛函下的吸附能的SISSO统一公式。(c)不同小分子与O*之间的描述符权重系数的线性标度关系。(d)不同泛函的描述符的权重系数。
论文由Guolin Cao, Shiyu He, Yirong Zhang, Sha Yang, Shuang Li, Yusheng Li, Ji-Chang Ren共同完成,第一作者为曹国琳,通讯作者为任吉昌副教授,通讯单位为南京理工大学。
3.总结与展望
该研究构建了一个兼具高精度与可解释性的多保真机器学习框架,实现了以极少量RPA 数据快速预测催化吸附能,并给出了明确的物理解析表达式。相关成果为高效催化材料的理论筛选提供了新工具,也为机器学习在材料科学中的“可解释建模”探索了新路径。未来,该方法有望拓展至高熵合金等复杂体系,加速新型催化材料的理性设计与发现。
4.文章链接
Guolin Cao, et al. “A data-driven framework for efficient and physically interpretable prediction of RPA-level adsorption energies” Materials Today Electronics 14 (2025) 100184
https://www.sciencedirect.com/science/article/pii/S2772949425000506

