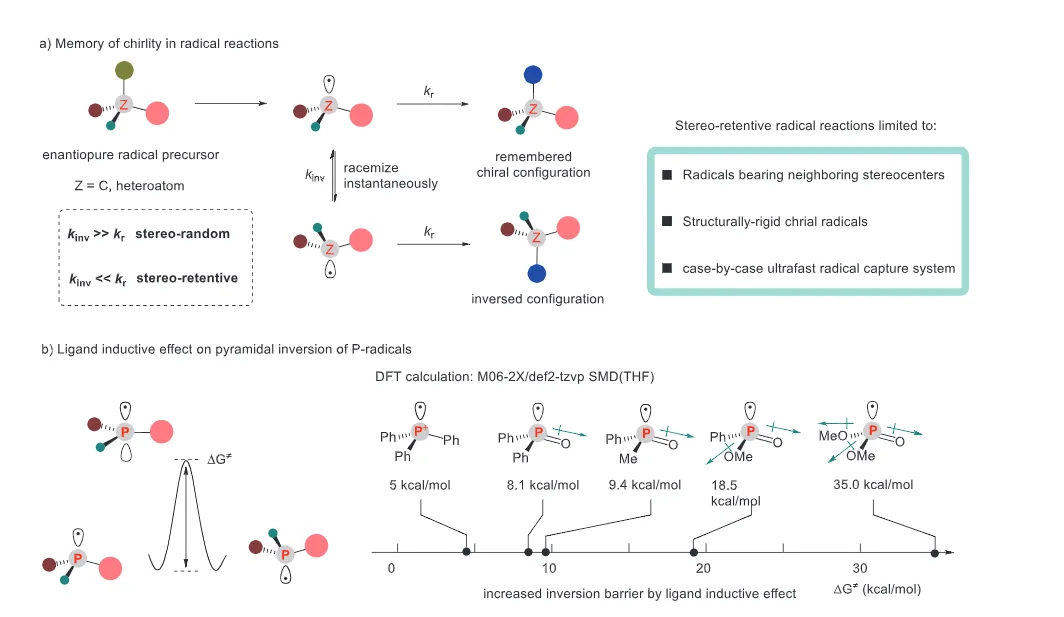
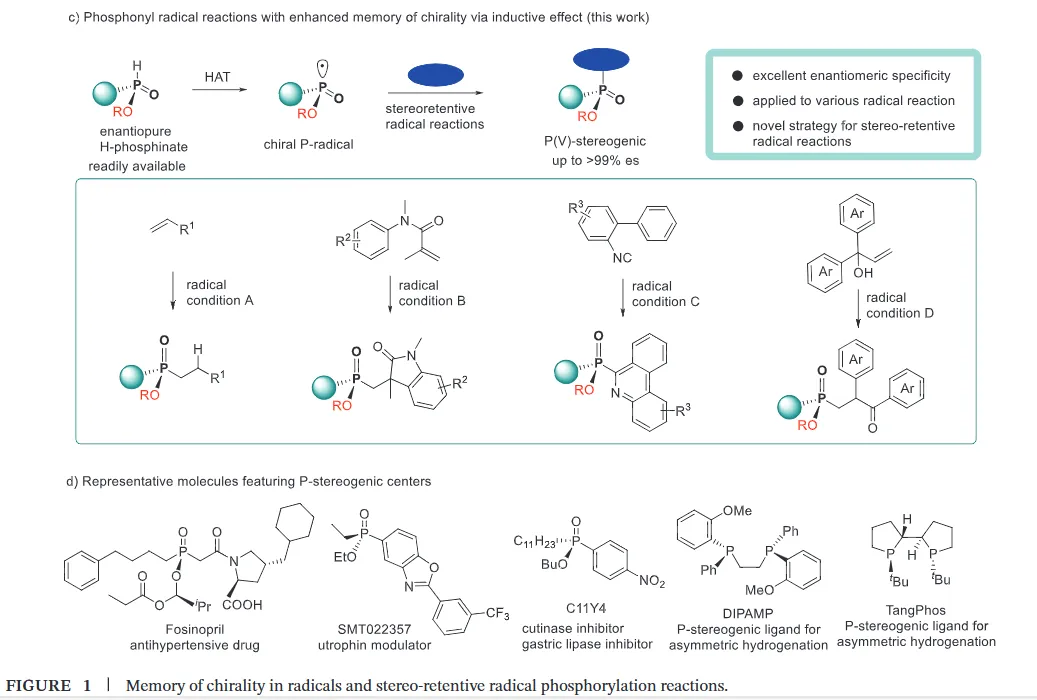
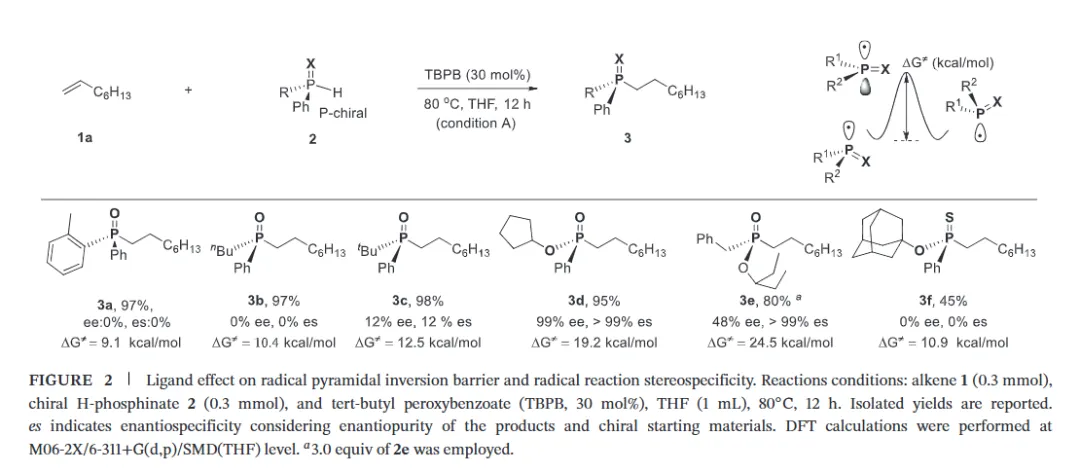
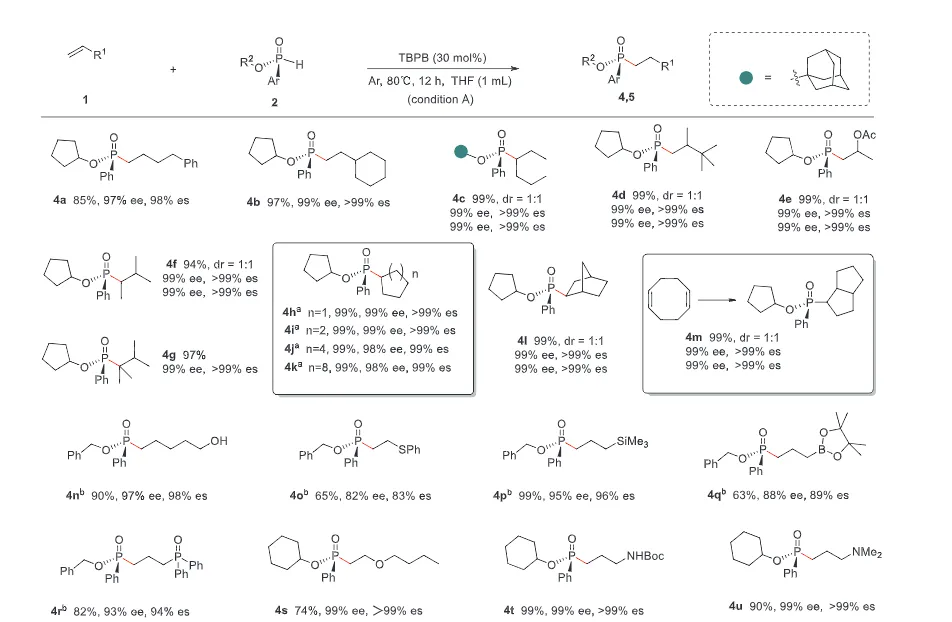
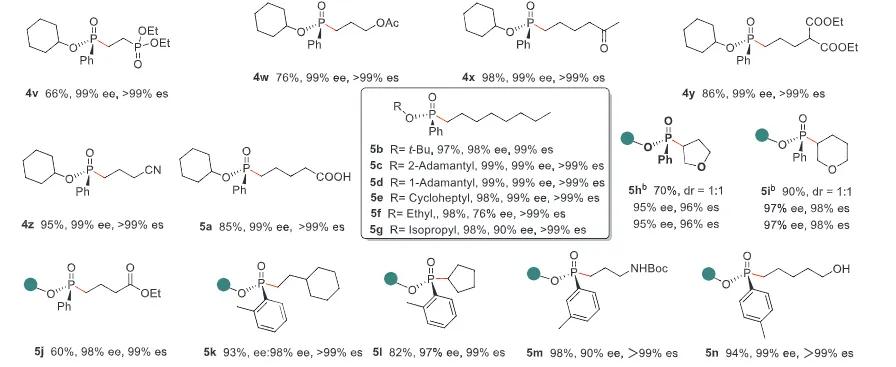
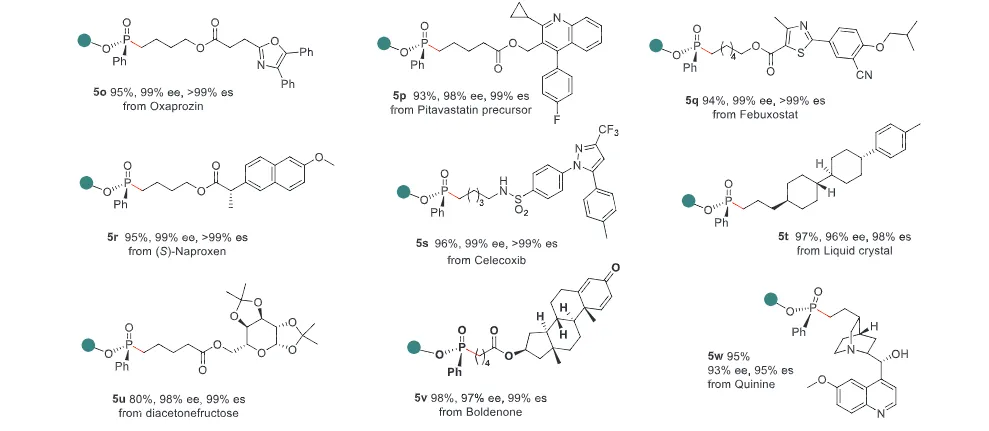
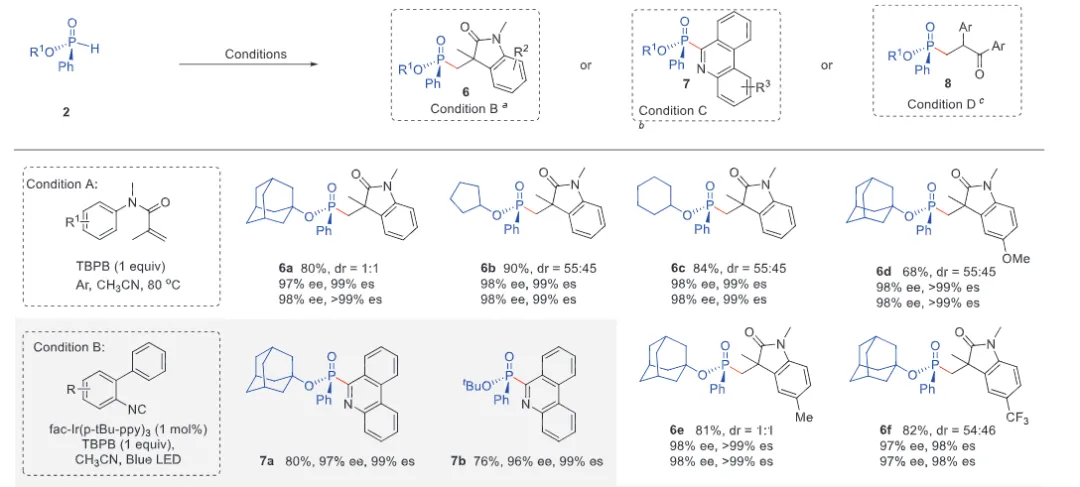
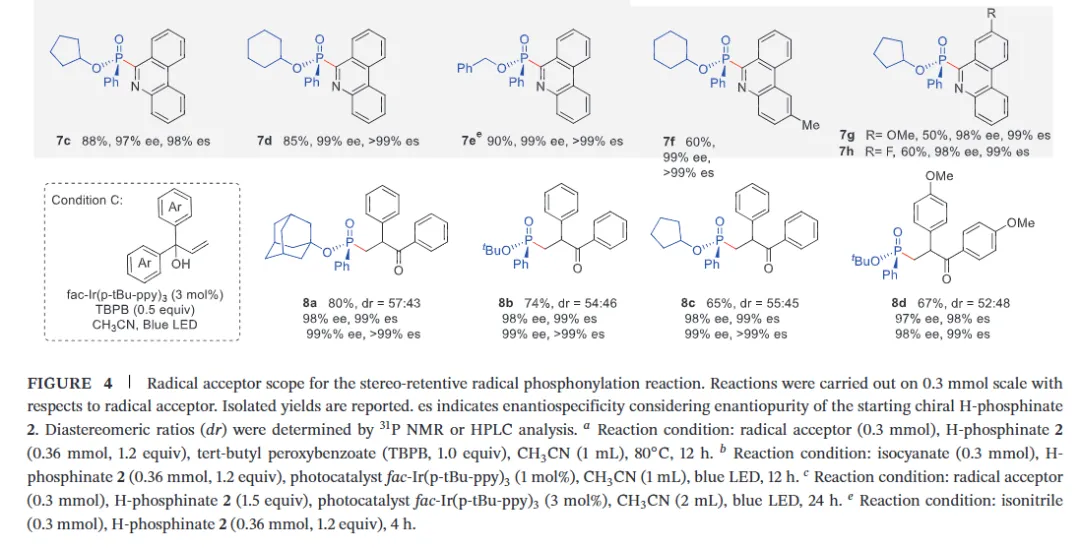
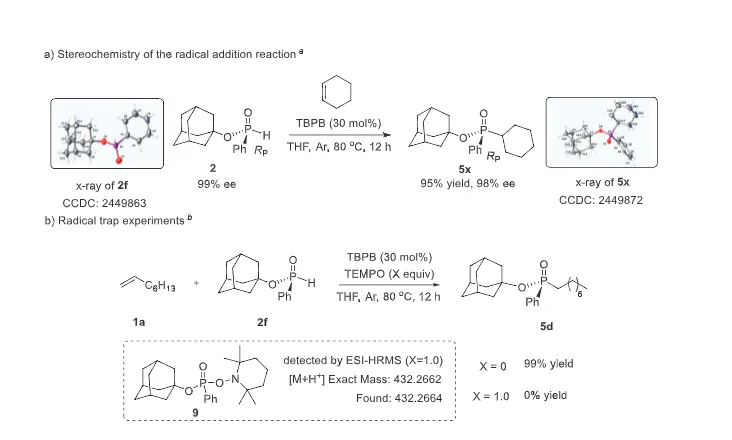
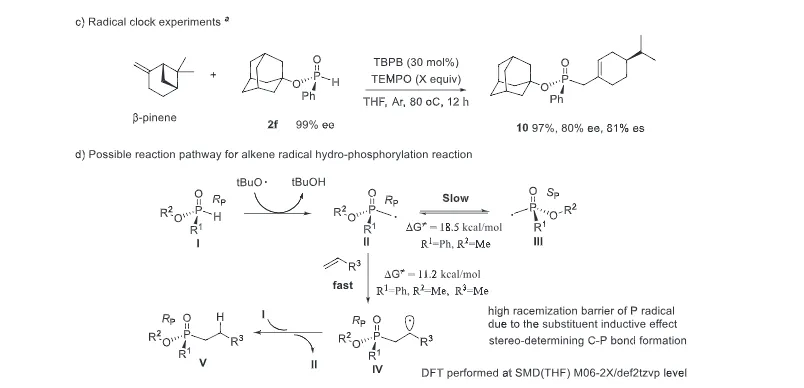
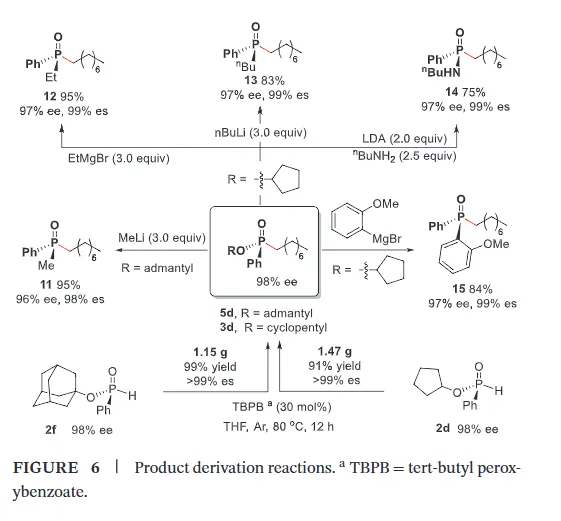
The realization of stereospecific radical reactions—particularly those involving heteroatom-centered radicals—remains aformidable challenge due to the rapid configurational inversion (racemization) of transient radical intermediates. This study presents a general strategy to enhance the memory of chirality (MOC) in radicals by exploiting the inductive effect of substituents. Through DFT calculations, we demonstrate that the pyramidal inversion barrier of phosphorus-centered radicals can be dramatically increased by substitution with highly electronegative atoms. This deceleration of inversion enables stereoretentive transformations of enantiopure H-phosphinates under mild, radical conditions. A series of stereospecificphosphoryl radical reactions, including alkene hydrophosphonylation, intramolecular arylphosphonylation, phosphorylation–cyclization of isocyanates, and aryl migration reactions, were successfully developed, providing access to diverse P(V)-stereogenic compounds with high efficiency (up to 99% yield) and excellent stereospecificity (up to >99% es). The utility of this approach is highlighted by the late-stage functionalization of densely functionalized pharmaceuticals, bioactive molecules, and a liquid crystal. This work establishes a foundational principle for achieving stereochemical control in radical reactions via rational tuning of theradical intermediate’s configurational stability.
图2. 配体效应对自由基棱锥形反转能垒和自由基反应立体选择性影响(图片来源于ACIE)图3. 立体选择性保留的自由基氢膦酰化反应底物拓展(图片来源于ACIE)图4. 立体选择性保留的自由基膦酰化反应自由基受体底物拓展(图片来源于ACIE)图5. 机理研究及可能的反应机理(图片来源于ACIE)