Epitaxial Active Interface to Construct Intralattice-Bonded Asymmetric Bi1─O─Bi2 Sites for Robust CO2 Photoreduction to Acetic Acid
构建外延活性界面以形成晶格内键合的不对称Bi1─O─Bi2位点,实现二氧化碳高效光还原制乙酸https://doi.org/10.1002/anie.202524970太阳能驱动下由二氧化碳(CO₂)选择性合成C₂化学品是碳循环的关键途径,但受制于C─C偶联反应的高动力学能垒。本研究提出了一种晶格键合不对称位点的外延生长策略,通过在Bi₃NbO₇纳米点/Bi₃O₄Br纳米片(BNO/BOB)界面构建Bi₁─O─Bi₂位点,促进C─C偶联以实现乙酸生成。该体系下CO₂光催化转化为乙酸的速率可达192.3 µmol·g⁻¹·h⁻¹,选择性达91.4%,380 nm和400 nm处的表观量子效率分别达9.49%和6.57%。其核心机制源于界面Bi₁─O─Bi₂位点触发的级联电子效应:界面电荷再分布诱导强内建电场,高能电子选择性占据CO中间体的2π反键轨道,显著削弱CO中间体中的C─O键;同时,不对称电荷再分布有效中和相邻CO中间体间的静电排斥,通过Bi位点的d-π电子反馈协同稳定OCCO过渡态。双效应协同降低了C─C偶联与加氢步骤的能垒,最终驱动反应路径向高稳定性乙酸生成方向进行。乙酸(CH₃COOH)是化学制造、食品加工和制药等行业广泛使用的重要化学品。2023年,全球乙酸需求量约达1927万吨,受生产需求增长的推动,预计到2030年将增至2360万吨。目前,甲醇羰基化法是乙酸合成的主导工艺,占全球产量的60%以上。然而,该工艺需高温高压条件,导致能耗显著。随着对能源消耗和环境影响的关注日益加剧,利用可再生资源开发可持续的CH₃COOH生产方法的需求愈发迫切。近期研究表明,太阳能驱动二氧化碳还原制乙酸可实现精细化学品的可持续生产,同时将温室气体转化为高附加值产品。该路径为环境友好型可持续化学制造提供了极具前景的方案。为推进该技术发展,开发能够促进二氧化碳光还原制乙酸的新型高效催化剂至关重要。
在二氧化碳光还原反应体系中,热力学与动力学限制通常导致C1产物(如CO、CH₄、HCOOH等)的生成。然而,更高价值且用途更广的C2产物(如CH₃COOH、CH₃CHO和CH₃CH₂OH)在实际反应中难以高效生成。这一难题主要源于催化剂表面C1中间体(如CO、CHO、*CH₃)的排列与相互作用优化能力有限。中间体间的偶极-偶极排斥显著限制了C─C偶联效率。近期研究表明,电荷非对称界面可显著降低C1中间体间的静电排斥,为C─C偶联创造更有利条件。因此,在合成CH₃COOH的光催化二氧化碳还原体系中,设计具有电荷非对称界面的光催化材料至关重要。异质结策略是一种有效途径,其中S型和Z型等电荷分离异质结通过能带工程构建内建电场,有效促进光生载流子的空间分离。然而,这些策略的界面功能主要局限于调控电荷流动方向。此外,传统复合体系中组分间物理化学性质的显著差异常导致相分离,使界面接触面积受限,阻碍电荷非对称界面的形成。相比之下,利用相同元素组成的基底构建外延异质结界面可形成更稳定的化学键,从而实现强界面耦合。此外,界面两侧不同晶相的存在可促进非对称位点的形成。
本研究成功在Bi₃NbO₇(BNO)纳米点表面实现了Bi₃O₄Br(BOB)纳米片的外延生长,构建了具有晶格键合非对称Bi₁─O─Bi₂位点的BNO/BOB异质结光催化剂。该催化剂乙酸生成速率达192.3 µmol·g⁻¹·h⁻¹,选择性为91.4%,380 nm波长下表观量子效率(AQE)达9.49%,显著优于已报道体系。研究表明,Bi₁─O─Bi₂界面键建立了超快电子传输通道并诱导界面电荷再分布,形成强内建电场驱动光生电子定向迁移。该机制可降低相邻CO*中间体间的静电排斥并促进C─C偶联,最终高效提升乙酸生成。
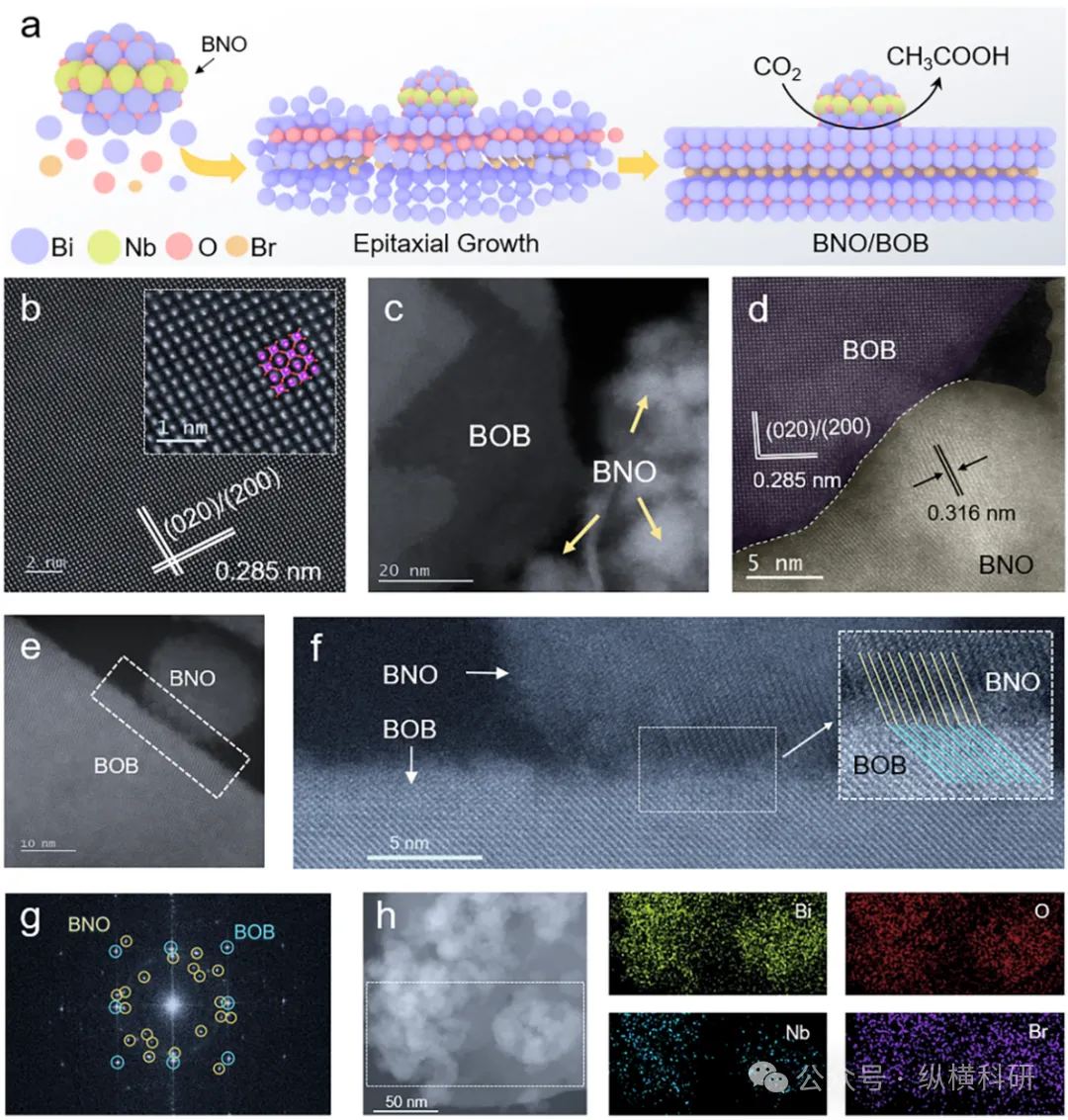
图1 (a) BNO/BOB形成过程示意图;(b) BOB的高角环形暗场扫描透射电子显微镜(HAADF-STEM)图像;(c - f) 7BNO/BOB的HAADF-STEM图像;(g) 7BNO/BOB的快速傅里叶变换(FFT)图谱;(h) 7BNO/BOB的扫描透射电子显微镜-能量色散X射线谱(STEM-EDS)面扫图像。
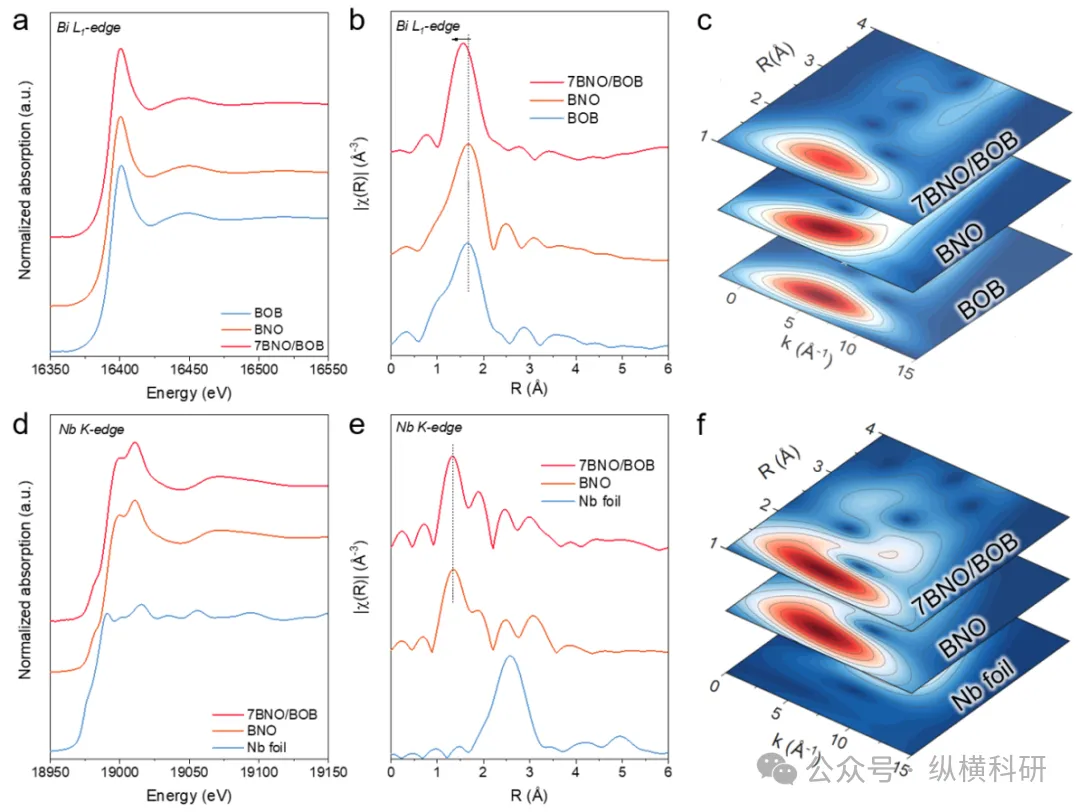
图2 BOB、BNO和7BNO/BOB在(a) Bi L₁边和(d) Nb K边的同步辐射X射线吸收精细结构(XAFS)光谱;BOB、BNO和7BNO/BOB在(b) Bi L₁边和(e) Nb K边的扩展X射线吸收精细结构(EXAFS)光谱;(c, f) 小波变换图。
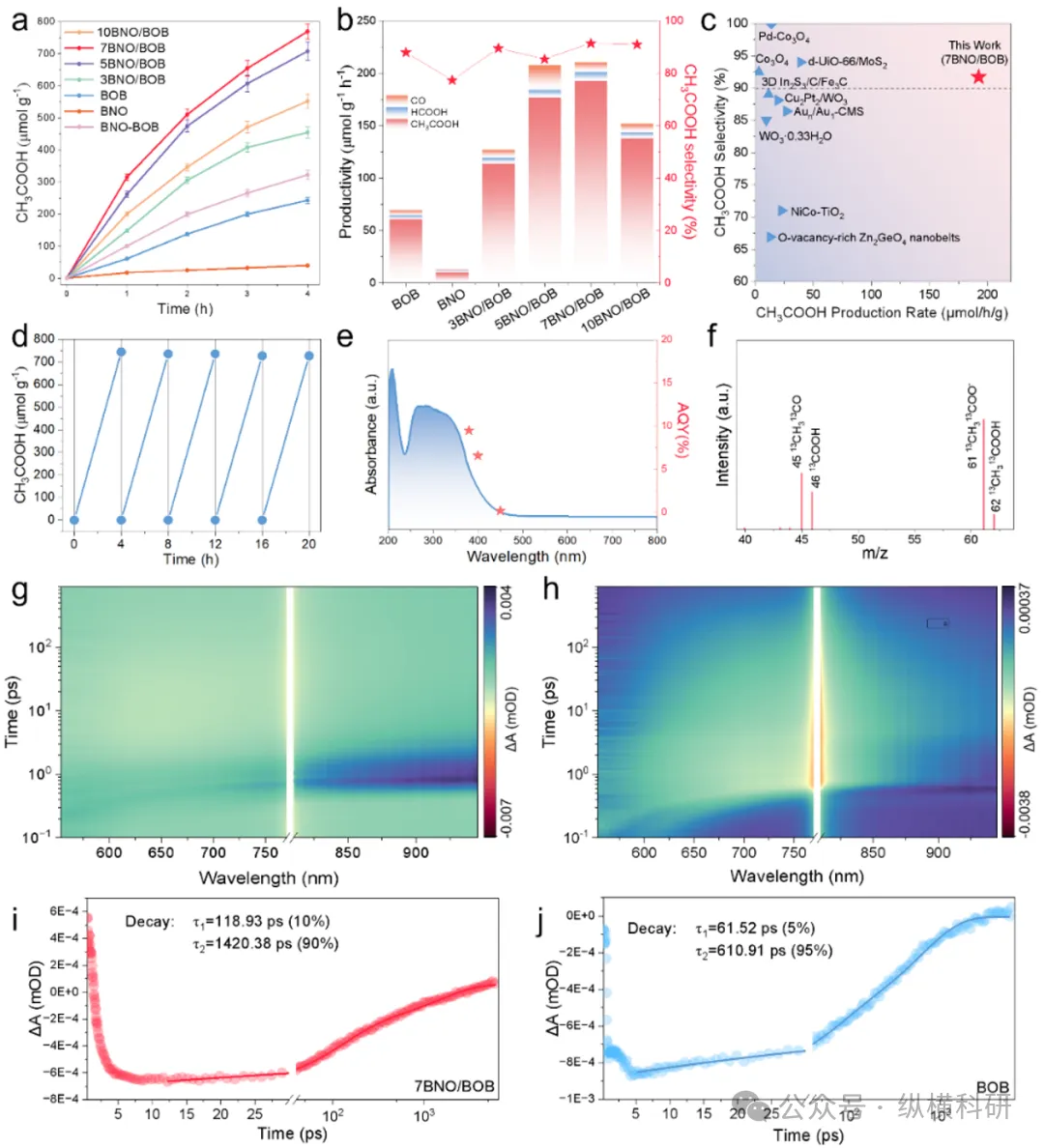
图3 (a) BNO、BOB、BNO/BOB和BNO - BOB光催化二氧化碳还原为乙酸的性能(误差棒代表至少三次独立实验的标准偏差);(b) BNO、BOB和BNO/BOB催化二氧化碳还原为乙酸的选择性;(c) 二氧化碳还原光催化剂的性能对比;(d) 7BNO/BOB催化二氧化碳还原的循环测试;(e) 7BNO/BOB的表观量子效率(AQE);(f) 7BNO/BOB催化¹³CO₂还原后产物的质谱图;(g) 7BNO/BOB的二维伪彩色瞬态吸收(TA)图;(h) BOB的二维伪彩色瞬态吸收(TA)图;(i) 7BNO/BOB的瞬态吸收衰减动力学;(j) BOB的瞬态吸收衰减动力学。
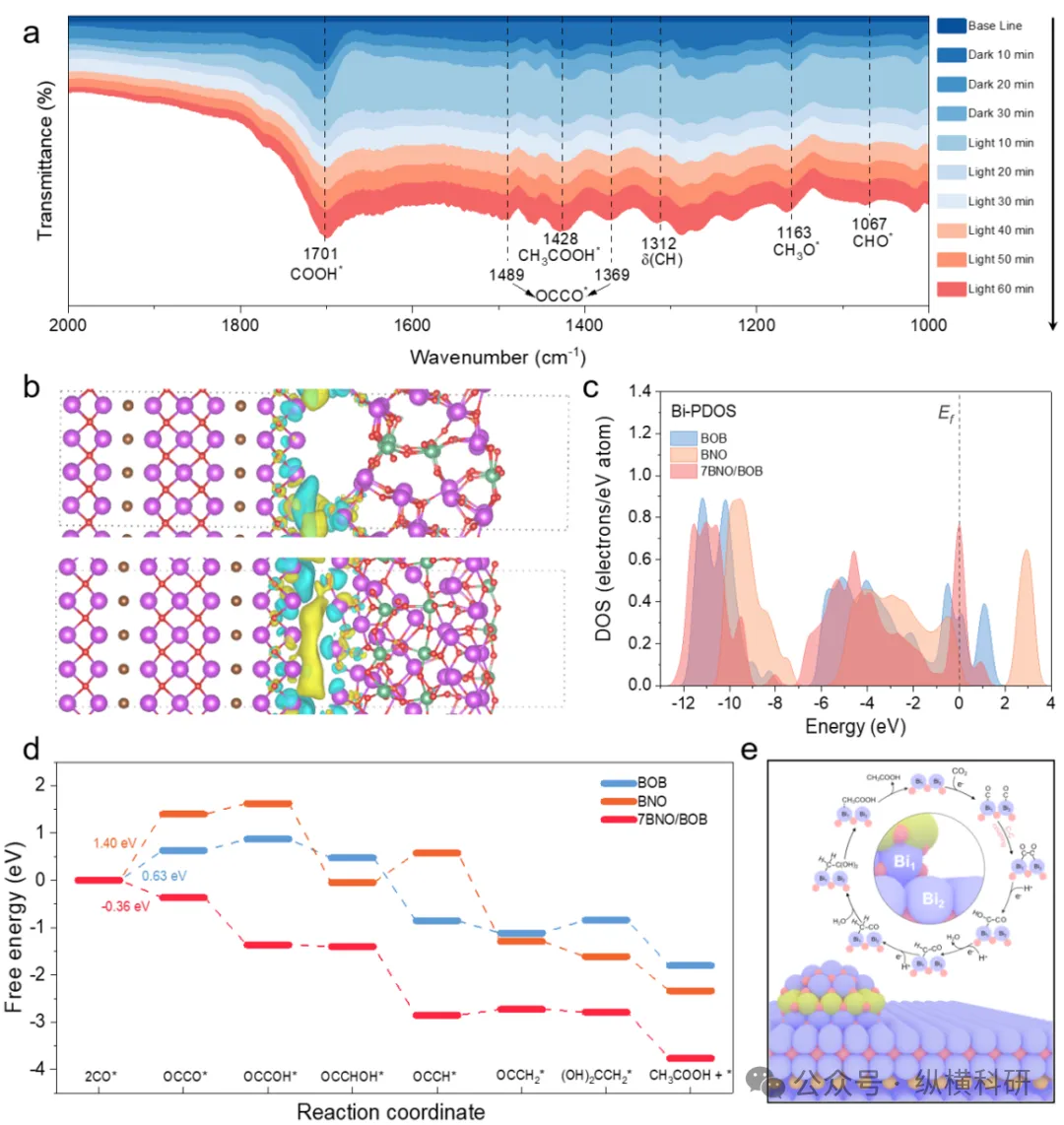
图4 (a) 7BNO/BOB上二氧化碳共吸附的原位傅里叶变换红外光谱(FTIR);(b) BNO/BOB界面的电荷差异图(电荷密度增加用黄色表示,减少用蓝色表示);(c) Bi的态密度(PDOS);(d) BOB、BNO和7BNO/BOB催化二氧化碳还原为乙酸盐的自由能图;(e) BNO/BOB光催化二氧化碳还原为乙酸的示意图。
综上所述,在Bi₃NbO₇(BNO)表面实现Bi₃O₄Br(BOB)的外延生长,可在BNO/BOB界面构建晶格内键合的非对称Bi₁─O─Bi₂位点,从而实现二氧化碳向乙酸的高效转化。综合表征与计算表明,Bi₁─O─Bi₂位点具有双重功能:既是电荷再分布引擎,又是关键中间体OCCO形成的促进剂。Bi₁─O─Bi₂作为界面超快电子传输通道,使电子高度富集并产生强内建电场。由此产生的高动能电子选择性占据CO的2π反键轨道,促使C─C偶联能垒显著降低至-0.36 eV。协同优化的电子环境同时降低了OCCO*的加氢能垒。最终,最优化的7BNO/BOB催化剂实现乙酸生成速率达192.3 µmol g⁻¹ h⁻¹(选择性91.4%),并在380 nm和400 nm波长下分别获得9.49%和6.57%的表观量子产率。本研究表明,通过构建晶格内键合非对称位点的原子尺度策略,可克服传统异质结中物理界面接触的局限性。该策略通过成键轨道调控电子迁移路径与反应微环境,为设计二氧化碳光还原制C₂⁺产物的催化剂提供了新的原子尺度设计范式。