大连理工大学&清华大学&南京理工大学&北京科技大学 Acta Materialia 通过双稳定纳米相的协同析出克服镁锂合金的高强不稳定性
- 2026-04-27 23:30:39
以体心立方(BCC)结构为主的镁锂铝合金因其卓越的比强度和延展性而备受关注。然而,固溶处理和水淬后形成的Mg3Al纳米析出相在室温下极易粗化,导致合金强度显著降低。为抑制这一不稳定性,本文提出一种合金成分优化策略,促使Mg3Al相转变为更稳定的半共格Mg(Al,Zn,Ag)Li2析出相,同时伴随界面处富锂团簇的共析出。为系统揭示该成分设计下DO3有序析出相的演化路径及其内在机制,本研究结合同步辐射X射线衍射和相场模拟进行了综合分析。第一性原理计算证实,Mg(Al,Zn,Ag)Li2相的形成焓低于Mg3Al相,表明其在室温下具有更高的稳定性。富锂团簇的存在有助于保持合金的BCC结构,并抑制基体向HCP结构的转变。分子动力学模拟结果表明,Mg(Al,Zn,Ag)Li2相与富锂团簇协同作用,在界面处诱发应力场,阻碍位错运动并增强位错间的相互作用,从而显著提升合金强度。同时,富锂团簇能够降低共格界面应力,减缓析出相粗化,有效补偿强度损失,使合金在高强度状态下保持优异的结构稳定性。

https://doi.org/10.1016/j.actamat.2026.122010
镁及其合金因其低密度和高比强度,成为航空航天和交通运输行业减重的理想候选材料。然而,镁合金在室温下塑性有限,这从根本上源于其密排六方晶体结构,该结构提供的独立滑移系远少于面心立方或体心立方晶格。虽然基面滑移因其临界剪切应力低而主导了大部分室温变形,但它仅能提供两个独立滑移系,导致无法容纳均匀塑性应变。激活额外的滑移模式是提高变形协调性的有效方法。稀土元素已被证明能够通过降低非基面滑移的临界剪切应力来激活这些滑移系,从而实现从单一基面滑移到协调的多滑移系变形的转变,以此提高合金的整体塑性。值得注意的是,改善镁合金室温成形性的一种更直接、更具变革性的方法是与锂合金化,这能从根本上改变基体结构。特别地,根据镁锂二元相图,约10.3 wt.%的锂含量是合金基体完全转变为BCC结构的临界点,从而显著提高拉伸延伸率。向BCC结构的转变标志着在克服镁合金室温成形性差的持久挑战方面取得了突破。尽管如此,这种二元镁锂合金的屈服强度通常较低,仍不足以满足工程应用对力学性能的严格要求,这极大地限制了它们的实际应用。
合金化在显著改善二元镁锂合金力学性能方面起着关键作用。值得注意的是,铝在镁锂合金中具有相当大的固溶度,不仅是固溶强化的理想选择,也适用于沉淀硬化。例如,Tang等人报道,他们开发的Mg-11Li-3Al-1(Zr, Y)合金在60分钟淬火后,压缩屈服强度超过350 MPa,与二元Mg-11Li合金(~85 MPa)相比,实现了约265 MPa的显著三倍增长。重要的是,这种显著的强度提升归因于淬火后10分钟内快速形成均匀分布、纳米棒状且半共格的D03-Mg3Al析出相,这些析出相作为高效的强化剂。然而,室温时效会诱发θ相的奥斯特瓦尔德熟化,同时伴随纳米级α-Mg颗粒在界面处析出。这种析出破坏了界面共格性并削弱了强化效果,导致整体软化。然而,铝引入的显著沉淀强化效应常常以牺牲塑性为代价。以LA113合金为例,偏聚在晶界的D03-Mg3Al相容易引发晶间分离,导致拉伸断裂应变低于2%,表现为典型的脆性断裂行为,严重限制了其工程应用前景。
为了克服合金化带来的脆性,研究人员探索了各种替代方法。一种策略是避免依赖脆性析出相,而是采用剧烈塑性变形工艺,例如对LA143合金进行多向锻造,以细化晶粒结构,实现超过80%的压缩断裂应变。另一种方法侧重于改性合金基体,例如利用Mg-7Li等双相合金,或采用高压处理和相细化来平衡强度和塑性。尽管在提高合金强度和塑性方面取得了显著进展,但合金在实际服役条件下保持性能稳定的能力仍然是工程应用面临的主要挑战。潜在微观结构的稳定性,特别是析出相的内在稳定性及其抗粗化能力,至关重要,但至今仍未得到充分理解。
析出相与基体之间的高度共格性对于决定合金高强度阶段的微观结构稳定性至关重要。新出现的证据表明,可控地增加锂含量可以降低基体的晶格参数(例如,从Mg-17Li的3.495 Å降至Mg-22.2Li的3.486 Å),从而有效减小析出相/基体界面处的晶格错配。通过减少晶格错配,可以最小化析出相与基体之间的界面应变能,从而有效减缓析出相的粗化动力学,进而提高强度稳定性。然而,大量的电化学测量和析氢实验表明,锂的高化学反应活性和电离趋势导致合金的腐蚀速率随锂含量增加而增加。相比之下,精确调控析出相的本征强度稳定性代表了一种更有效的策略,然而这种方法鲜有报道。尽管先前的研究报道了合金中如MgAlLi2(D03有序结构,Fd3m,a = 6.37 Å)和AlLi(B32有序结构,Fd3m,a = 6.37 Å)等析出相的形成和晶体结构,但对其本征稳定性,特别是它们与界面处共析出的富锂团簇之间的相互作用和协同效应的系统而深入的理解仍然有限。同时,虽然人工时效诱导亚稳态Mg3Al相向稳定AlLi相转变可以提高合金的强度稳定性,但这仍然会导致整体强度显著降低。这种退化是因为AlLi相与基体非共格且数密度低,因此无法提供有效的强化。因此,引导Mg3Al相向着与基体保持共格/半共格关系的更稳定析出相转变,对于实现此类合金高强度与室温稳定性之间的平衡具有关键价值。
在本研究中,我们成功诱导了半共格且热力学更稳定的纳米Mg(Al,Zn,Ag)Li2相与更细小的富锂团簇的协同析出,这有助于确保此类合金在微观结构和强度方面具有出色的长期稳定性。所开发的LAZQ11233合金展现出最高的比强度(348.68 kN·m/kg),可与马氏体时效钢相媲美,同时其强度稳定性与高强度镁锂铝合金相比提高了近2.5倍。我们不仅通过实验实现了这种相变路径,还结合第一性原理计算和分子动力学模拟,从电子和原子尺度揭示了这种析出相的稳定性机制及其强化效应。本研究提出的相变策略为开发兼具高强度和稳定性的镁锂合金提供了关键的理论和实验基础,具有重要的科学和工程意义。
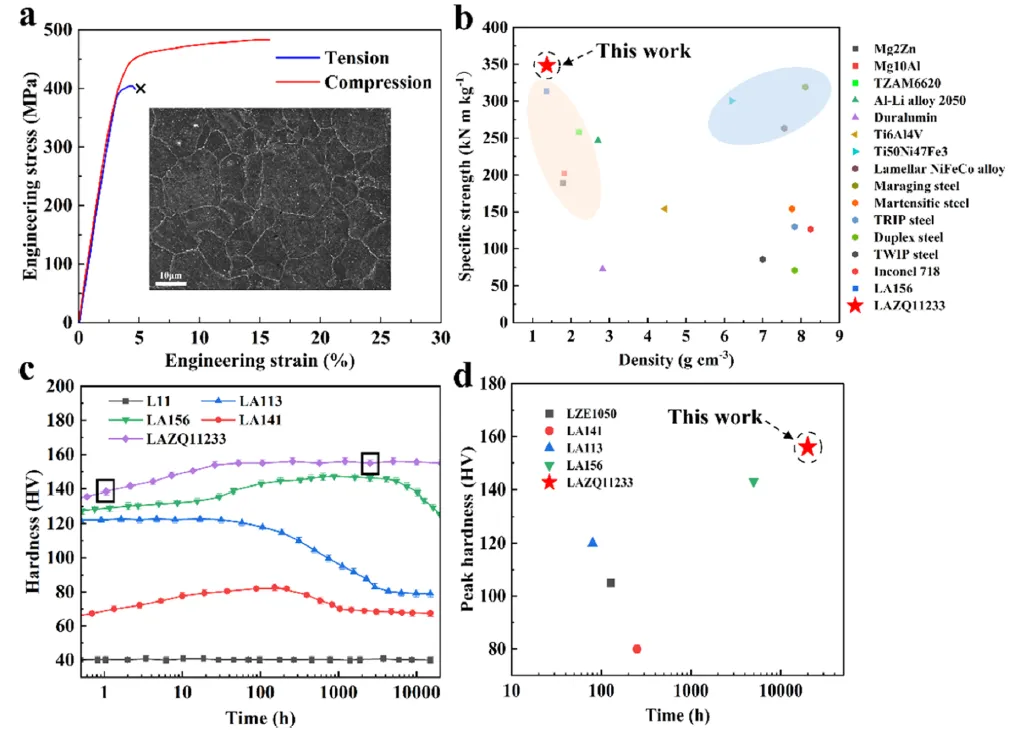
图 1. LAZQ11233 合金的力学性能。(a) 淬态 LAZQ11233 合金的应力-应变曲线(插图为代表性显微组织)。(b) LAZQ11233 合金与一系列高强度合金(包括镁基、铝基合金、钢和高温合金)的比屈服强度对比。(c) LAZQ11233 合金及其他一系列 BCC 镁锂合金的硬度随时间变化曲线。(d) LAZQ11233 合金与其他 BCC 镁锂铝或镁锂锌合金的峰值硬度保持时间对比。

图 2. 室温时效 LAZQ 合金的微观结构。 透射电子显微镜图像、高角环形暗场-扫描透射电子显微镜图像的代表性数据。(a) LAZQ 合金的 HAADF 图像(室温时效 1 小时)。(b) 对应(a)图的衍射花样。红色方框突出显示(110)β 和(200)β 晶面,黄色方框分别标记(200)β 和(111)β 晶面。(c) 高分辨率图像及相应的快速傅里叶变换花样。右上角和右下角的插图分别对应蓝色方框标记的区域(标记为 FFT#1 和 FFT#2)。(d) (c)图中红色方框区域的逆快速傅里叶变换图像,显示了析出相-基体界面(白色虚线)。红色和蓝色点分别代表推断的超晶格和基体原子位置。(e) 对应(a)图中 HAADF 图像的 EDS 元素面分布图,显示了 Mg(红)、Zn(紫)、Al(绿)和 Ag(黄)的空间分布。
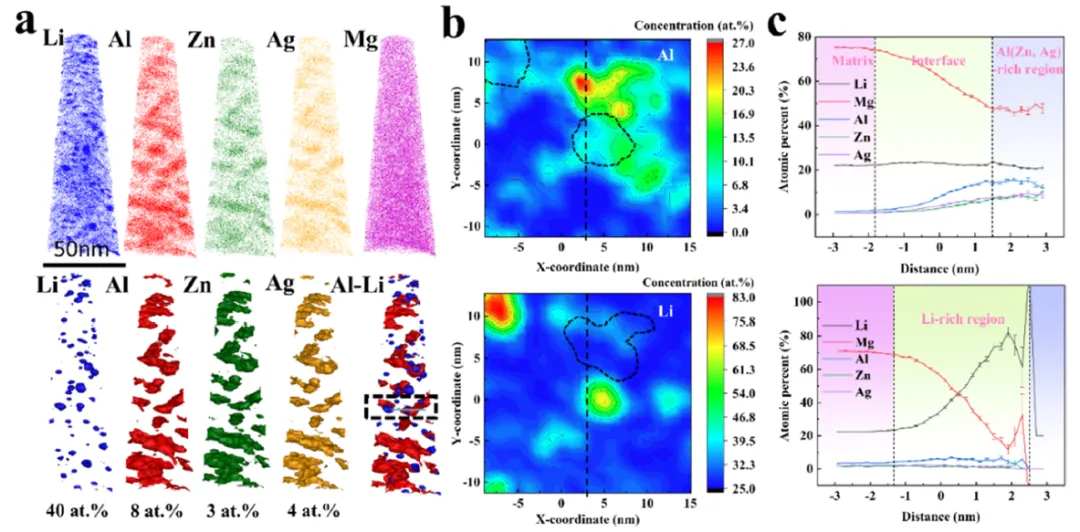
图 3. 原子探针层析技术分析。(a) 40 × 40 × 100 nm³ 体积的 APT 重构图像,显示了 Li、Al、Zn、Ag、Mg 的分布。插图分别对应以 40 at.% Li、8 at.% Al、3 at.% Zn 和 4 at.% Ag 等浓度面突出的析出相。(b) 感兴趣区域(图 3a 中黑色方框区域)内 Al 和 Li 的 APT 浓度分布图,突出显示了 θ 相核心区域的 Li 贫化以及富锂团簇内同时发生的 Al 贫化(由黑色虚线方框强调)。析出相的锂铝比约为 2,并在相邻基体区域内弥散分布着富锂团簇。(c) 近邻图显示了通过(Al, Zn, Ag)富集析出相时的成分变化。界面宽度约为 3.3 nm,富锂团簇的尺寸约为 4 nm。
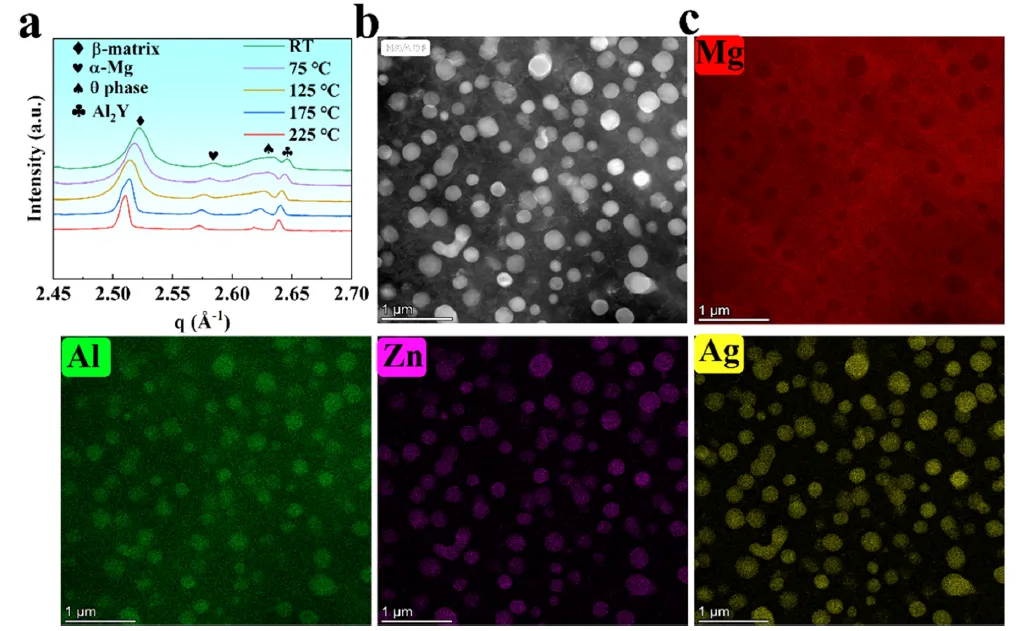
图 4. LAZQ11233 合金的原位高温同步辐射 WAXS 图谱及微观结构表征。(a) 室温至 225°C 范围内的 WAXS 图谱(注:q = 4πsinθ/λ,其中 θ 是散射角,λ 是入射 X 射线波长)。对应于 Mg(Al, Zn, Ag)Li₂ 相的衍射峰在 175°C 开始出现,并在 225°C 时呈现轻微窄化并向低 q 值方向移动,这是由于适度的晶粒长大和热膨胀引起的晶格参数变化所致。(b) LAZQ11233 合金的 HAADF 图像,显示了析出相的形貌和微观结构特征(室温时效 5016 小时,对应图 1c 中左侧黑色方框)。(c) 对应(b)图中 HAADF 图像的 EDS 元素面分布图,显示了 Mg(红)、Zn(紫)、Al(绿)和 Ag(黄)的空间分布。
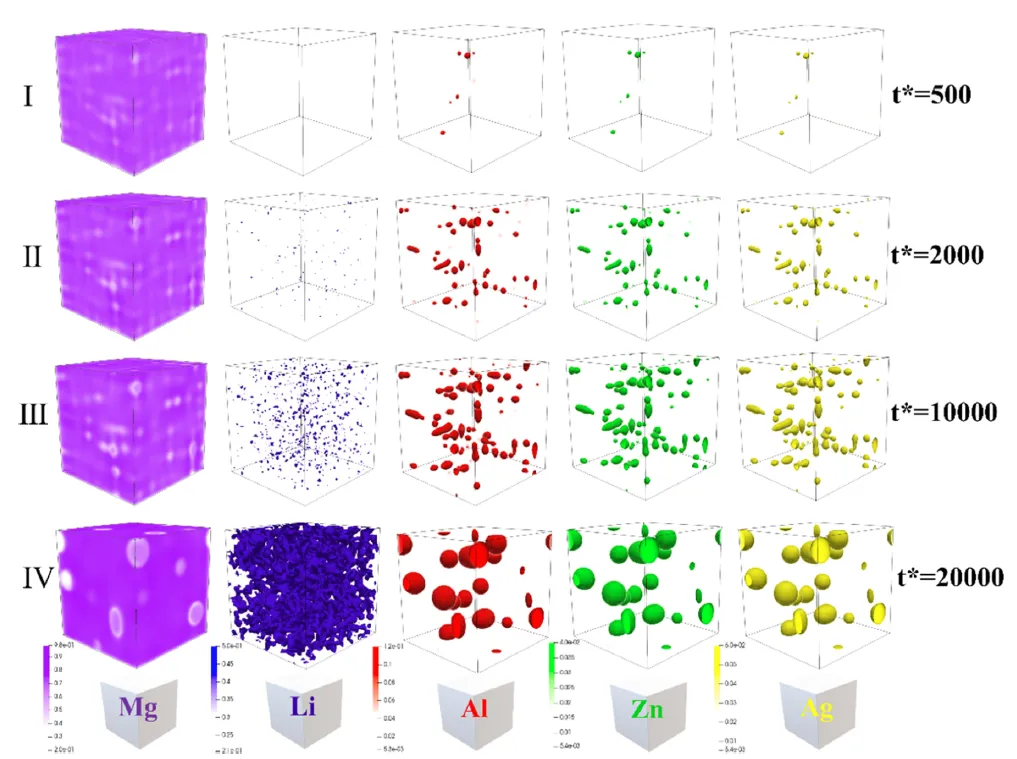
图 5. LAZQ11233 合金中相变的相场模拟结果。 Mg(Al, Zn, Ag)Li₂ 相和富锂团簇的微观结构演化。主要阶段包括:I,形核;II,定向粗化;III,平衡;IV,奥斯特瓦尔德熟化。序参量(任意单位)定义在 0 到 1 的范围内,表示从无序固溶体向完全有序 D0₃-Mg(Al, Zn, Ag)Li₂ 相的转变。
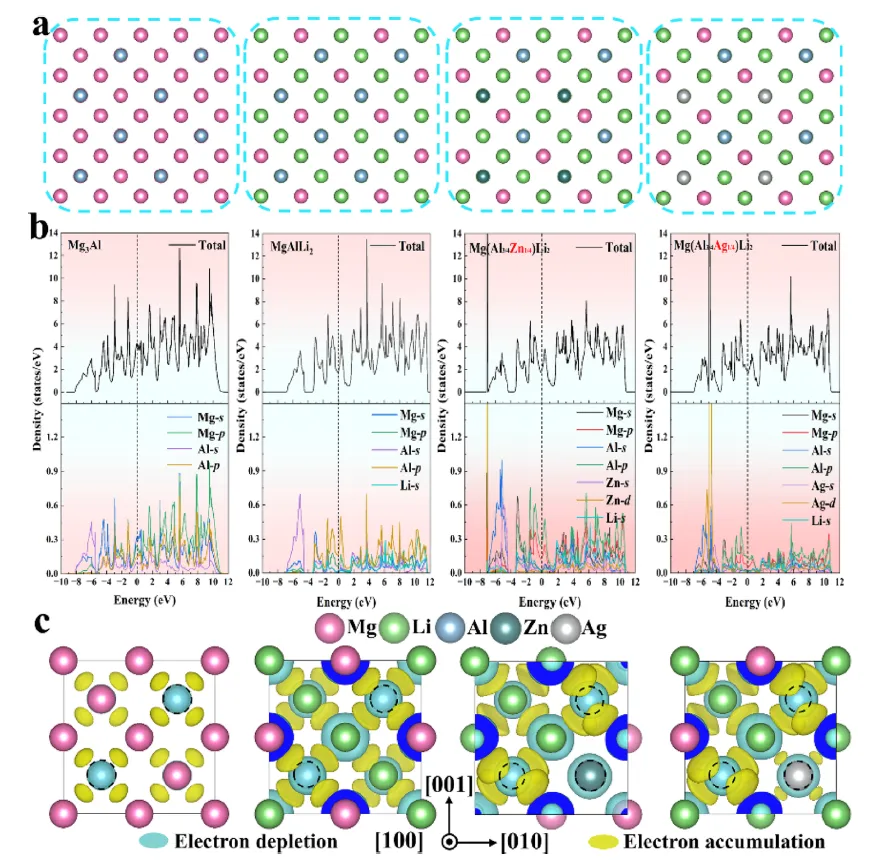
图 6. 基于第一性原理计算的 Mg₃Al 及 Zn/Ag 掺杂模型的原子结构和电子性质。(a) 纯 Mg₃Al、MgAlLi₂、Mg(Al₃/₄Ag₁/₄)Li₂ 和 Mg(Al₃/₄Zn₁/₄)Li₂ 的原子结构。(b) Mg₃Al、MgAlLi₂、Mg(Al₃/₄Ag₁/₄)Li₂ 和 Mg(Al₃/₄Zn₁/₄)Li₂ 的总态密度/分波态密度。费米能级设为零。Zn 和 Ag 的引入导致费米能级附近的电子结构发生显著改变,从而影响键合特性和相稳定性。采用了基于广义梯度近似的密度泛函理论计算。(c) Mg₃Al、MgAlLi₂、Mg(Al₃/₄Ag₁/₄)Li₂ 和 Mg(Al₃/₄Zn₁/₄)Li₂ 的差分电荷密度分析。等值面(水平 = ±0.003 e⁻/ų)描绘了电子聚集(黄色)和耗散(青色)区域。
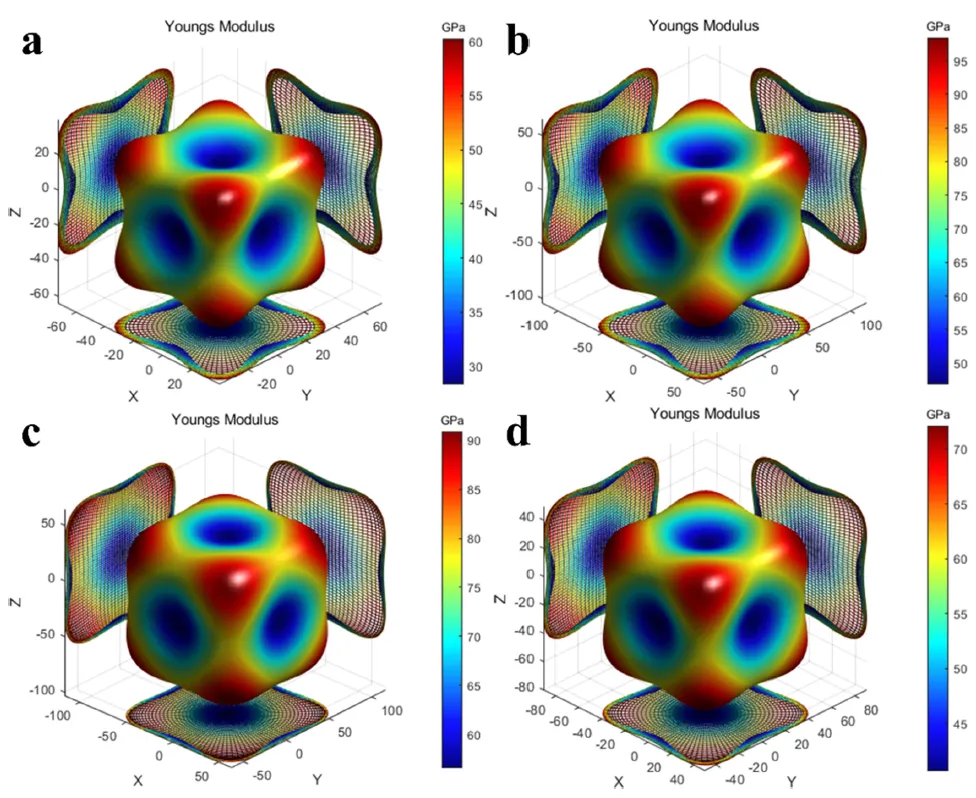
图 7. 通过第一性原理计算得到的 Mg₃Al 及其 Zn/Ag 掺杂结构的三维杨氏模量。(a) Mg₃Al。(b) MgAlLi₂。(c) Mg(Al₃/₄Zn₁/₄)Li₂。(d) Mg(Al₃/₄Ag₁/₄)Li₂。模量在 <100> 方向上达到最小值,在 <111> 方向上达到最大值。沿 <100> 方向的这种弹性软化促进了析出相在该方向上的择优生长,以最小化共格应变能。
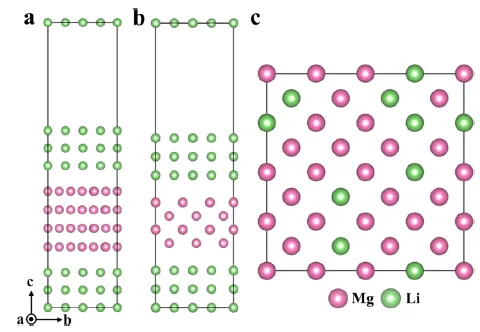
图 8. 基于第一性原理计算构建的镁锂体系原子模型。(a) β-Li (BCC) / α-Mg (HCP) / β-Li (BCC) (BCC-HCP-BCC) 结构。(b) β-Li (BCC) / α-Mg (BCC) / β-Li (BCC) (BCC-BCC-BCC) 结构。(c) Li 在 Mg 中的无序固溶体模型,显示了 Li 在 BCC 镁基体中的随机分布。
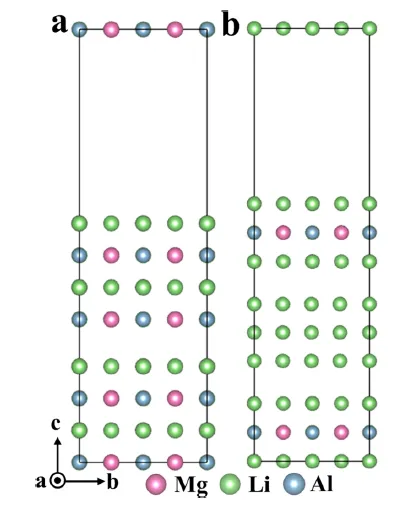
图 9. 基于第一性原理计算构建的镁锂铝体系与锂之间的异质界面原子模型。(a) MgAlLi₂/MgAlLi₂。(b) MgAlLi₂/Li/MgAlLi₂。
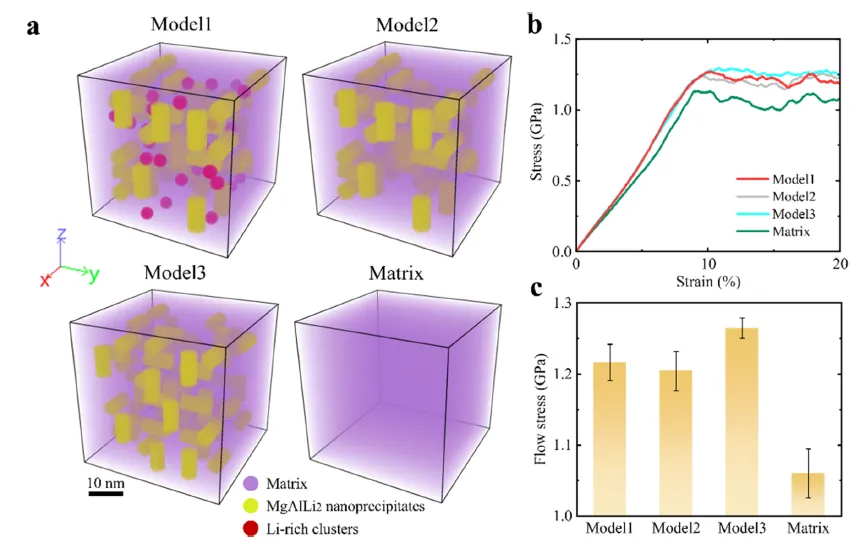
图 10. 通过原子模拟捕捉的塑性变形动态过程。(a) 三个模拟样品及镁锂基体的原子构型。基体、MgAlLi₂ 和富锂团簇中的原子分别以紫色、黄色和红色显示。(b) 单轴压缩的应力-应变曲线。(c) 所有模拟样品之间流变应力的比较。
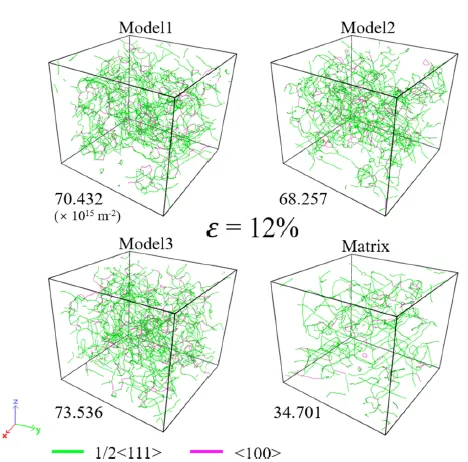
图 11. 不同模拟样品在 12% 应变下的位错结构。
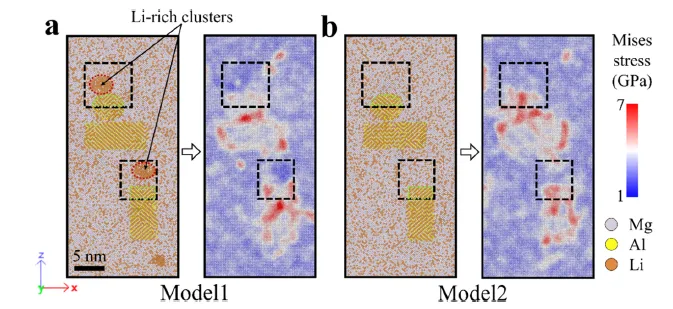
图 12. 模型 1 和模型 2 在 10% 压缩应变下的 MD 快照和米塞斯应力分布的切片视图。 Mg、Al 和 Li 原子分别以紫色、黄色和红色显示。
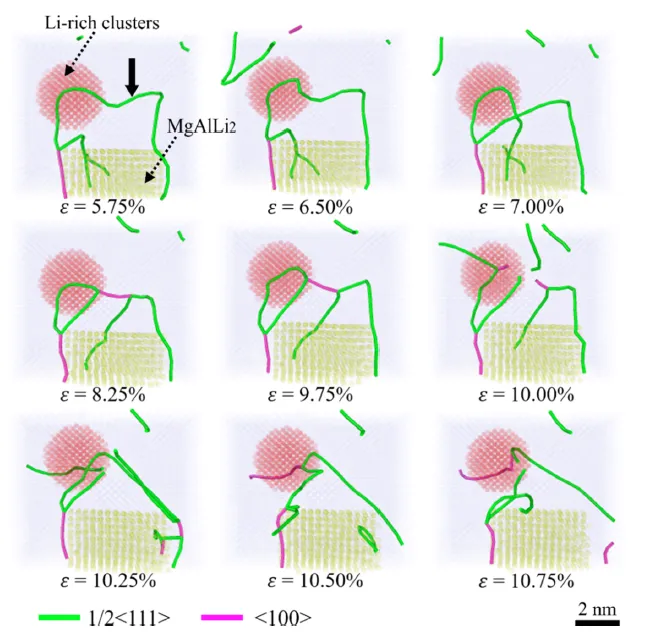
图 13. 模型 1 的 MD 模拟序列快照。 这些快照显示了位错、MgAlLi₂ 纳米析出相和富锂团簇之间复杂的相互作用。
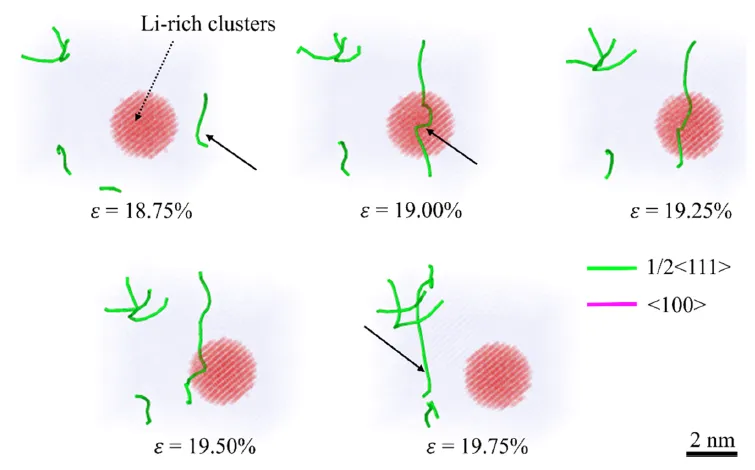
图 14. 位错与富锂团簇之间的相互作用。 显示位错切过富锂团簇的序列快照。
合金设计与制备:
研究团队设计并制备了一种新型多组分镁锂铝合金,命名为LAZQ11233。其名义成分为Mg-11.05Li-2.21Al-3.13Zn-2.97Ag-0.15Zr-0.61Y(wt.%)。
通过引入Zn和Ag元素,取代传统合金中的单一Al元素,旨在调控析出相的演化路径。
力学性能突破:
LAZQ11233合金展现出卓越的室温力学性能:拉伸屈服强度约395 MPa,比强度高达348.68 kN·m/kg,可与马氏体时效钢媲美,但密度降低了近四分之三。
更为关键的是,其强度稳定性得到了革命性提升。该合金在室温下能保持峰值硬度长达10,000小时,比目前报道的高强LA156合金提高了2.5倍以上,成功解决了传统Mg-Li-Al合金在室温下快速软化的痛点。
微观结构表征:
利用高分辨透射电子显微镜、原子探针层析技术等手段,系统表征了合金的微观结构。
关键发现:在时效后的合金中,并未观察到传统的不稳定Mg
3 3 2 更令人瞩目的是,在该析出相与基体的界面附近,发现了大量弥散分布的、尺寸约4 nm的富锂团簇。这是该合金微观组织的一个核心特征。
多尺度理论模拟:
结合第一性原理计算、相场模拟和分子动力学模拟,从电子结构、热力学、动力学到位错行为,全面揭示了材料强化的本质。
文章的核心在于揭示了Mg(Al,Zn,Ag)Li
1. 析出相稳定性提升机制:从亚稳到稳定
问题根源:传统Mg-Li-Al合金中的Mg
3 解决策略:通过添加Zn和Ag,它们会参与析出反应,将Mg
3 2 电子结构证据:第一性原理计算表明,Mg(Al,Zn,Ag)Li
2 3
2. 富锂团簇的作用:稳定基体与界面
稳定BCC基体:虽然合金的整体Li含量足以形成BCC结构,但APT发现析出相周围的基体Li含量较低。理论上,该区域应发生BCC向HCP的转变,但实际并未发生。DFT计算揭示,界面处析出的富锂团簇产生的应力场抑制了基体的HCP相变,维持了有利于塑性的BCC结构。
缓解界面应力:分子动力学模拟显示,在无富锂团簇时,析出相与基体界面存在严重的应力集中。而富锂团簇的存在能有效降低界面的应力集中,使得析出相与基体的界面更加共格、稳定,从而延缓了析出相的粗化动力学。
3. 协同强化机制:阻碍位错运动
双相协同:分子动力学模拟揭示了Mg(Al,Zn,Ag)Li
2 过程解析:位错在运动中首先遇到富锂团簇,部分位错切入团簇,部分位错被钉扎。随后,这些被阻碍的位错与后续位错发生复杂反应,形成位错环和不可动位错段。析出相和团簇共同导致位错发生显著的弓出、缠结和相互作用,极大地增加了位错运动的阻力,从而产生强烈的强化效应。
补偿强度损失:模拟还证实,尽管Mg(Al,Zn,Ag)Li
2
该研究通过多组分合金化策略,成功诱导了Mg(Al,Zn,Ag)Li

