我们建立了催化交流群,感兴趣的老师可以添加微信。微信号:nyhhjch001期刊信息:Science Advances, 2026, 12: eaeb1299
论文题目:Asymmetric Ru-O sites in self-activated catalysts for efficient electrochemical methanol oxidation and industrial-scale hydrogen generation
第一作者:Yujun Guo、Xueqin Mu
通讯作者:Suli Liu、Dingsheng Wang、Zhihui Dai
通讯单位:南京工业大学化学与分子工程学院;清华大学化学系;南京师范大学化学与材料科学学院等
全文速览
常规电解水制氢中,阳极 OER 动力学缓慢且产物氧气价值有限,导致整体能耗偏高。用甲醇氧化反应 MOR 替代 OER,可以在降低制氢电压的同时,在阳极联产甲酸盐 HCOO-。但要让 MOR 在工业级电流密度下同时具备高活性、高选择性和长寿命,仍然需要稳定的 Ru 基界面结构。
本文构建了RuOx@Mo(Mn)Ox催化剂:RuOx 纳米颗粒锚定在缺陷丰富的 Mo(Mn)Ox 纳米片上,并形成不对称 Ru-O-M(M = Mo 或 Mn)界面位点。该界面可以促进电子转移、稳定高活性 Ru 物种,并优化 MOR 中间体吸附。
性能上,该催化剂在三电极体系中实现 1000 mA cm-2 @ 1.41 V vs. RHE,甲酸盐法拉第效率超过 98%;两电极 MOR || HER 电解槽在 1000 mA cm-2 下电压仅 1.58 V,并稳定运行超过 300 h;AEMWE 中在 1000 mA cm-2 下电压为 1.87 V,可稳定运行 42 h,展示了甲醇氧化辅助制氢的工业化潜力。
研究背景
电解水制氢的主要能耗瓶颈来自阳极 OER。OER 不仅过电位高,而且生成的氧气附加值有限。将 OER 替换为小分子氧化反应,是降低电解槽电压、提高经济性的有效策略。其中 MOR 可将甲醇转化为甲酸盐,同时阴极仍进行 HER 产氢,因而具备“制氢 + 化学品联产”的优势。
问题在于,MOR 在高电流密度下容易受到界面电荷传输、活性位稳定性和反应中间体选择性的限制。Ru 基材料活性较高,但 Ru-O 键过度氧化和晶格氧机制可能引起结构衰退。本文的核心思路是通过 Mo(Mn)Ox 基底和 RuOx 之间的不对称 Ru-O 界面,既提高电子传输效率,又稳定 Ru 活性中心。
研究内容
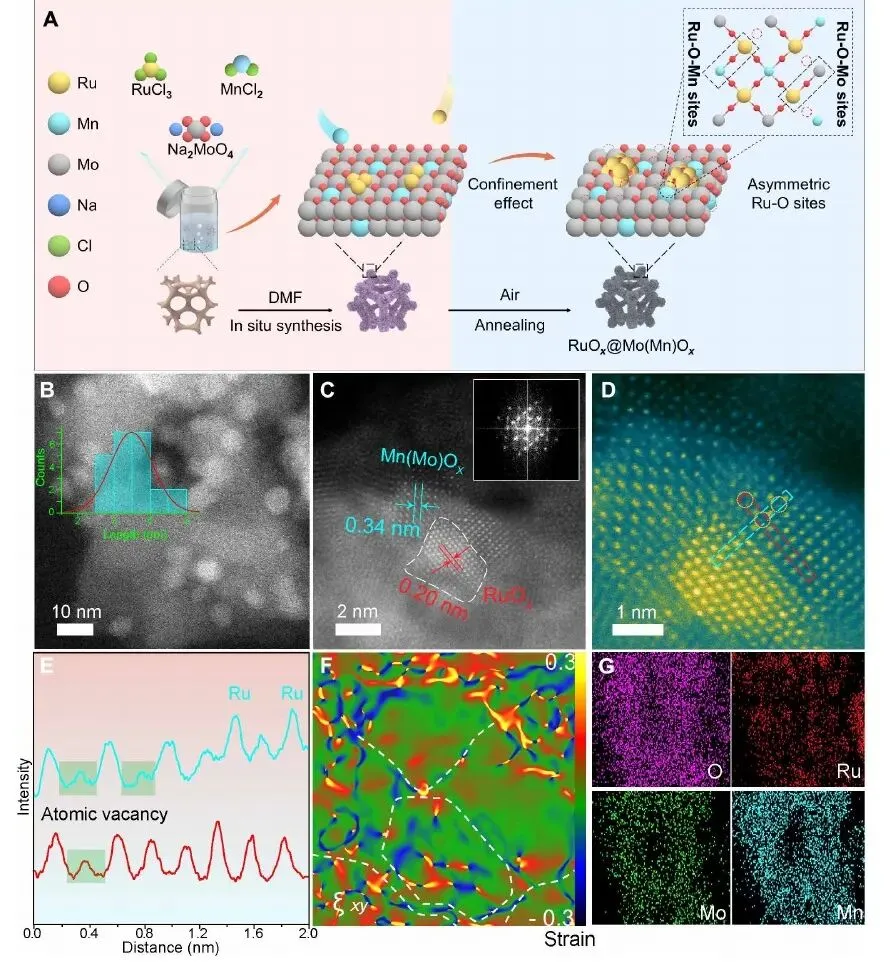
图 1. RuOx@Mo(Mn)Ox 的合成示意图、HAADF-STEM、晶格条纹、原子强度、应变分布和元素映射。
图 1 展示了催化剂结构设计。作者以 RuCl3、MnCl2 和 Na2MoO4 为前驱体,经原位合成和空气退火得到 RuOx@Mo(Mn)Ox。RuOx 纳米颗粒被限制并锚定在 Mo(Mn)Ox 纳米片网络上,形成三维片层堆叠结构。
HAADF-STEM 显示 RuOx 颗粒尺寸约 3.5 +/- 0.5 nm,HRTEM 中 0.20 nm 晶格条纹对应 RuO2 (101) 面,0.23 nm 晶格条纹对应 Mo(Mn)Ox (101) 面。原子强度和 GPA 应变图揭示界面处存在明显缺陷和晶格畸变,这些局部不饱和配位有利于形成不对称 Ru-O-M 位点。元素映射进一步证明 Ru、Mo、Mn、O 分布均匀,说明该结构不是简单颗粒混合,而是具有连续界面的复合氧化物网络。
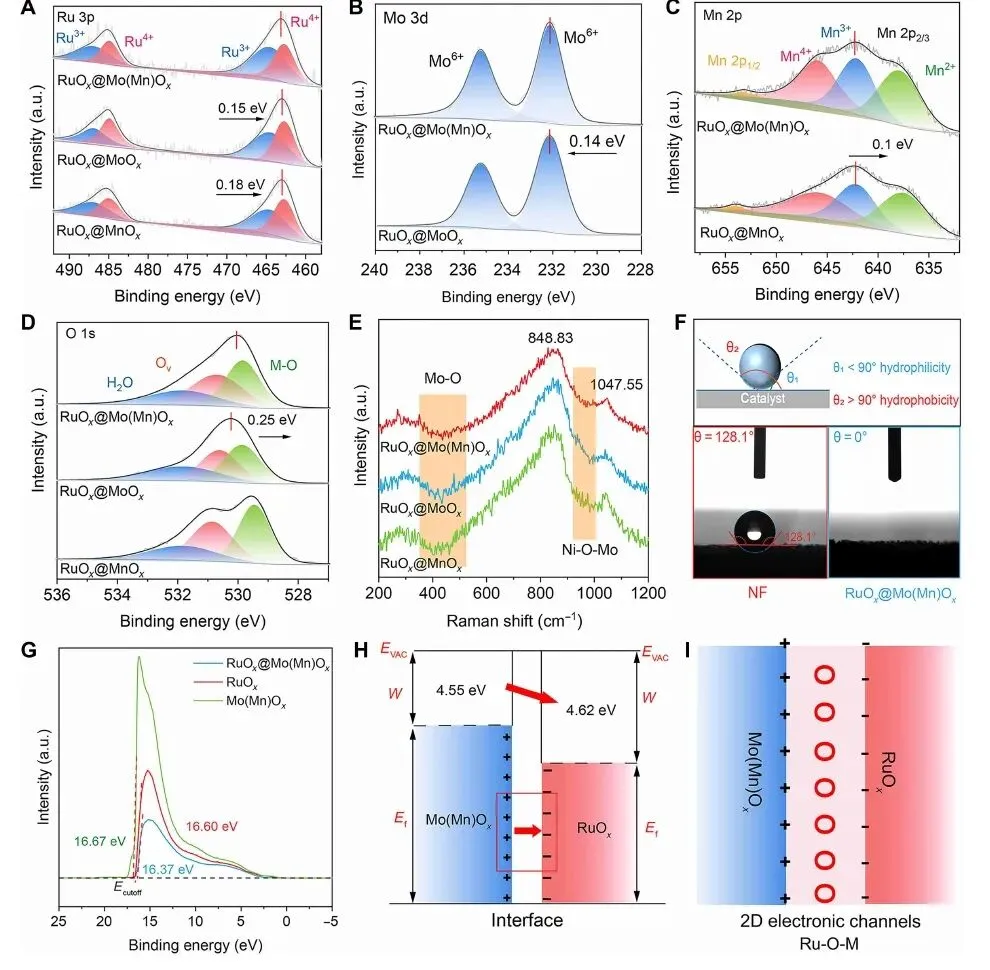
图 2. Ru 3p、Mo 3d、Mn 2p、O 1s XPS,Raman、接触角、功函数和界面电荷转移示意图。
图 2 说明不对称界面确实改变了 Ru 的电子结构。Ru 3p XPS 中 Ru3+ 与 Ru4+ 共存,比例约为 1:1,说明 Ru 位点处于混合价态,有利于电子转移和催化动力学。Mo 3d、Mn 2p 和 O 1s 峰位偏移则表明 Mo(Mn)Ox 与 RuOx 之间存在明显电子相互作用,界面氧参与构建 Ru-O-M 电荷传输通道。
Raman 中 Mo-O 和 Ni-O-Mo 相关信号进一步支持复合结构的形成。接触角测试显示 RuOx@Mo(Mn)Ox 表面接触角接近 0°,远优于裸 Ni 泡沫的 128.1°,说明其对含甲醇 KOH 电解液具有更好润湿性。功函数分析显示电子由 Mo(Mn)Ox(4.55 eV)向 RuOx(4.62 eV)转移,并使复合材料功函数升至 4.83 eV,反映出强界面偶极和电子重构。
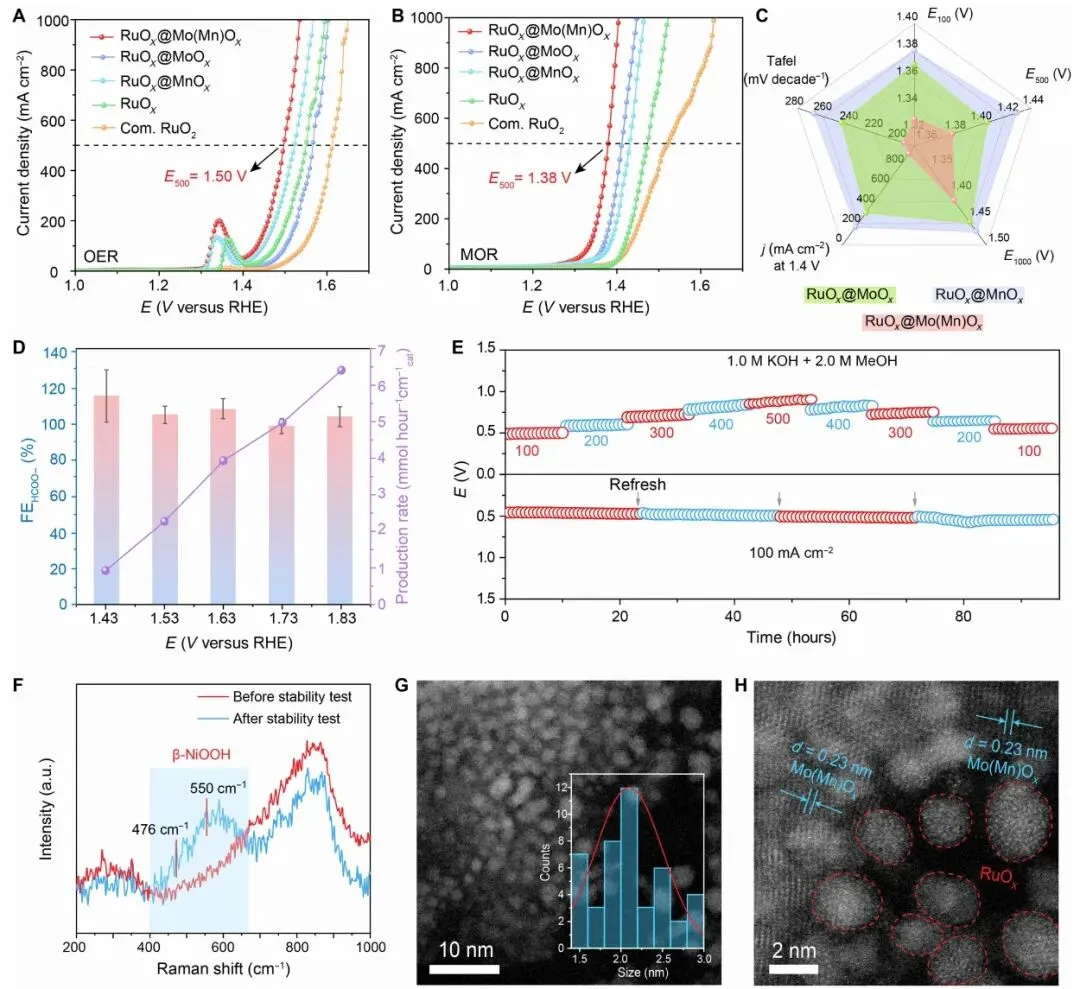
图 3. 1.0 M KOH 及 1.0 M KOH + 2.0 M MeOH 中的 OER/MOR 性能、甲酸盐 FE、产率、稳定性和反应后结构表征。
图 3 直接比较了 OER 与 MOR。未加入甲醇时,RuOx@Mo(Mn)Ox 在 500 mA cm-2 下对应电位约 1.50 V;加入 2.0 M 甲醇后,达到同一电流密度所需电位降至 1.38 V,较 OER 低 120 mV。该样品在 1.40 V 时电流密度达到 877 mA cm-2,明显高于 RuOx@MoOx 的 329 mA cm-2 和 RuOx@MnOx 的 184 mA cm-2。
产物选择性同样突出:在 1.43-1.83 V 范围内,HCOO- 法拉第效率超过 98%,且产率随电位升高而增加。恒电流测试中,催化剂可在 100-500 mA cm-2 扰动电流下稳定工作,并在 100 mA cm-2 下进行长期测试。反应后 Raman 出现 β-NiOOH 相关峰,HAADF-STEM 显示 RuOx 结构仍保持,颗粒尺寸约 2.2 +/- 0.1 nm,证明界面结构在 MOR 条件下具有较好稳定性。
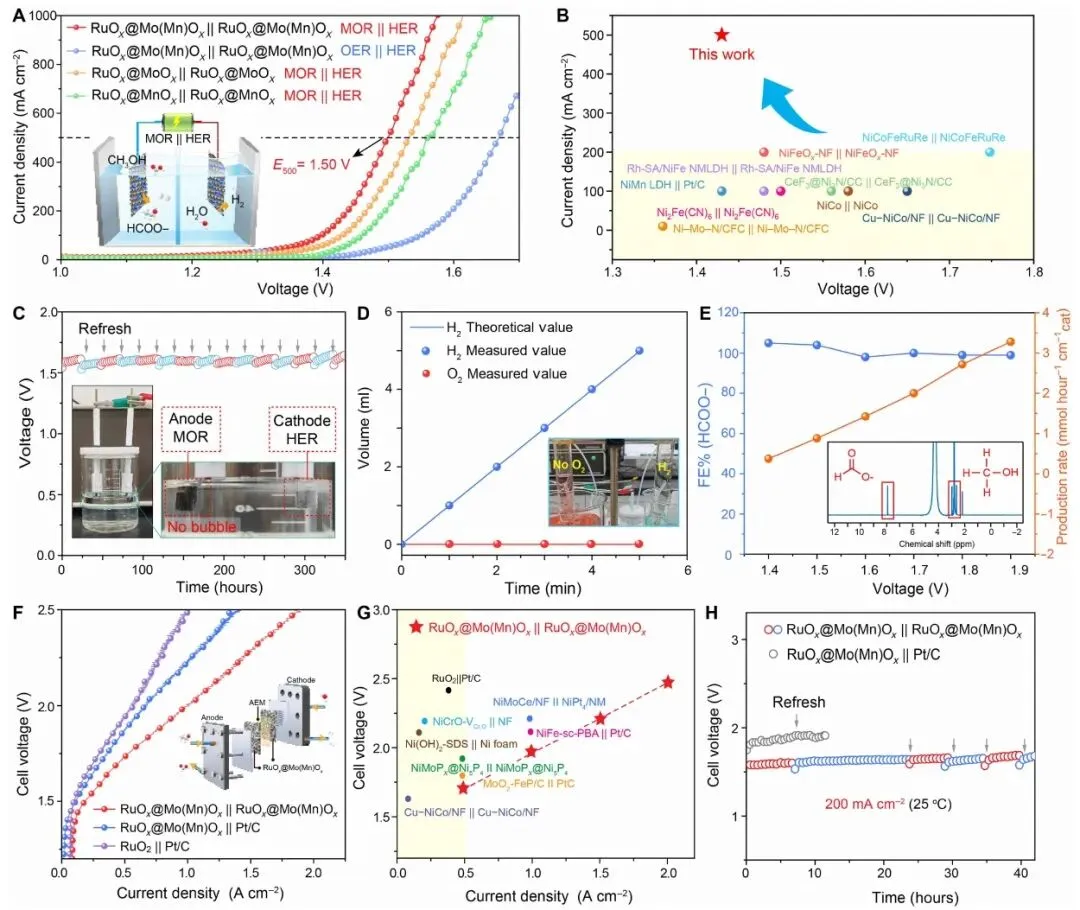
图 4. MOR || HER 两电极体系、产气/产物选择性、AEMWE 极化曲线、性能对比和稳定性测试。
图 4 将催化剂放入更接近应用的两电极体系。RuOx@Mo(Mn)Ox || RuOx@Mo(Mn)Ox 在 MOR || HER 条件下,100 mA cm-2 所需电压为 1.40 V;在 500 mA cm-2 时,相比常规水电解,加入甲醇后电压由 1.67 V 降低 170 mV。该体系在 100 mA cm-2 下经历超过 300 h 运行并周期补液,电压保持稳定。
产物测试显示,阴极 H2 产量接近理论值,而阳极 O2 基本被抑制,说明 MOR 有效替代了 OER。HCOO- FE 在 1.4-1.9 V 范围内超过 95%。在 AEMWE 中,该催化剂作为阴阳极均可工作,在 1000 mA cm-2 下电压为 1.87 V;在 200 mA cm-2 下稳定运行 42 h,且优于 RuOx@Mo(Mn)Ox || Pt/C 对照体系。
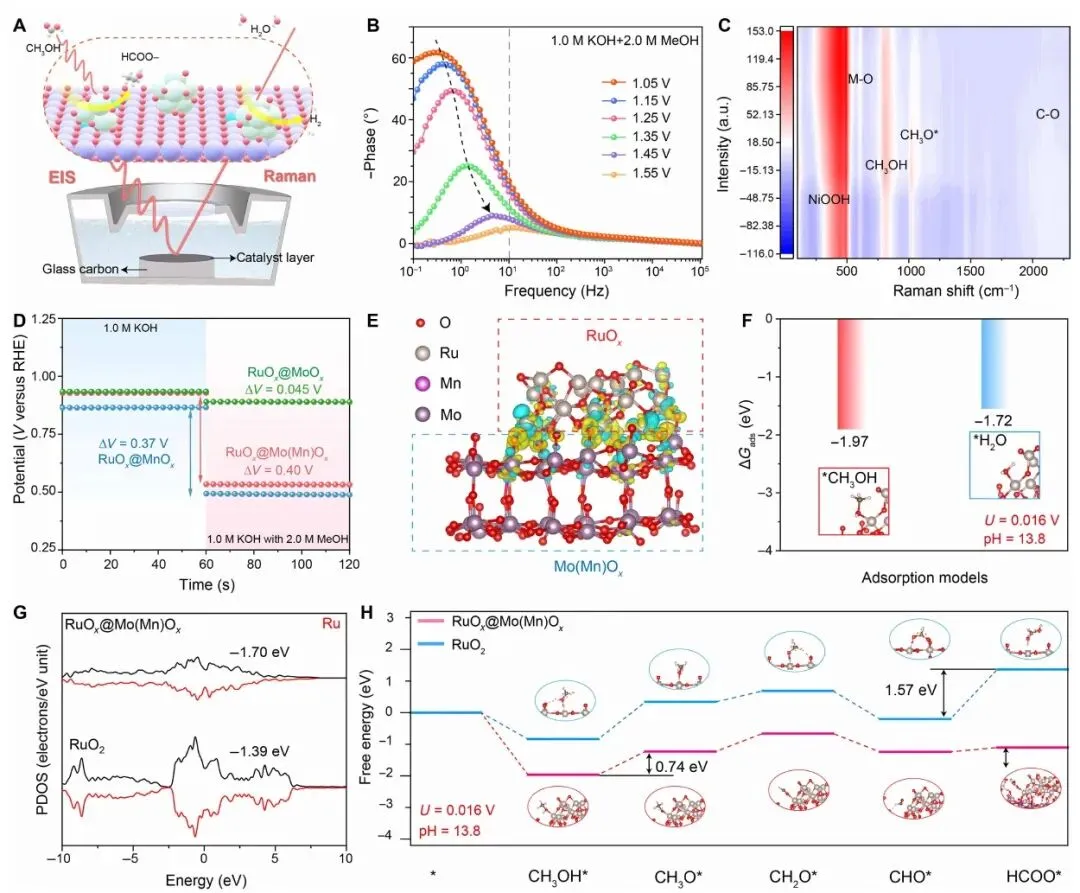
图 5. 原位 EIS/Raman 机制示意、MOR Bode 图、原位 Raman、OCP、界面结构模型、吸附能、PDOS 和 MOR 自由能路径。
图 5 解释了性能提升的机制。原位 EIS 显示,在 1.05-1.55 V MOR 电位窗口中,Bode 图相位角随电位升高明显降低,说明界面电荷传输阻抗减小,MOR 中多中间体转化过程被加速。原位 Raman 观察到 NiOOH、CH3OH、CH3O* 和 C-O 等相关信号,说明甲醇氧化中间体在工作电位下逐步形成和转化。
OCP 测试显示加入甲醇后 RuOx@Mo(Mn)Ox 电位变化最大,表明其对甲醇吸附/活化更敏感。DFT 结果显示,相比 RuO2,RuOx@Mo(Mn)Ox 对 CH3OH* 的吸附更有利;MOR 自由能路径中,RuO2 的限制步骤为 CHO* 到 HCOO*,能垒 1.57 eV,而 RuOx@Mo(Mn)Ox 的限制步骤转变为 CH3OH* 到 CH3O*,能垒降至 0.74 eV。这表明不对称 Ru-O-M 界面可以稳定关键中间体并降低甲醇氧化能垒。
结论与展望
本文通过界面工程构建了具有不对称 Ru-O-M 位点的 RuOx@Mo(Mn)Ox 催化剂。Mo(Mn)Ox 基底提供缺陷和电荷调控环境,RuOx 提供高活性氧化位点,二者之间的界面偶极和电子转移通道使 Ru 位点在高电流 MOR 条件下保持活性与稳定。
从应用角度看,该工作把甲醇氧化替代 OER 的优势推进到工业级电流密度:不仅显著降低制氢电压,还能高选择性联产甲酸盐。对于后续有机小分子辅助电解制氢而言,这篇文章提示界面电荷通道、活性位稳定化和产物选择性需要被同时设计,而不是只追求低过电位。
原文链接
https://doi.org/10.1126/sciadv.aeb1299


