南京师范大学孙瀚君教授ACS Catal.:d¹⁰构型金属介导的不对称吸附构型Co-OH-M(d¹⁰)缓解OH堵塞问题以实现高效海水电解析氧
- 2026-06-01 02:57:25

论文信息

第一作者:王申、张嘉晨
通讯作者:孙瀚君
通讯单位:南京师范大学
背景
钴基磷化物、硫化物和硒化物等材料(Co-Xides, X=P、S、Se)是极具潜力的耐腐蚀海水电解阳极催化剂,这源于其在反应过程中原位重构形成的含氧阴离子层(如PO₄³⁻、SO₄²⁻、SeO₄²⁻)能通过静电排斥作用有效抑制氯离子吸附。然而,在重构形成的OER活性相CoOOH中,关键氧中间体OH通过对称的d-p-d轨道构型Co-OH-Co被过度稳定在活性位点上,导致活性位点堵塞,显著抬升了后续OER步骤的能垒。基于此,本文通过引入具有d¹⁰构型的元素(In,Sn,Sb)成功构建了不对称的d-p-p轨道构型Co-OH-M(d¹⁰)。该结构利用轨道能级与对称性上的失配效应,显著降低了催化剂表面的OH覆盖度并促进了活性位点的循环再生。得益于此,具有代表性的Sn-CoOOH-PO₄³⁻催化剂分别在模拟海水和碱性天然海水中,于750 mA cm⁻²(1.78 V)和500 mA cm⁻²(2.05 V)的电流密度下分别稳定运行1000 h和500 h,展现出了出色的活性与耐久性。综上,本工作突破了耐氯腐蚀与高效析氧之间的传统权衡关系,为设计高效的海水电解催化剂提供了一个新的思路。
图文解析
(1)催化剂的设计原则
首先通过理论计算对上述催化剂设计思路的可行性进行了分析。以Sn作为d¹⁰构型金属的代表,构建了PO₄³⁻修饰的CoOOH与Sn-CoOOH模型(表示为CoOOH-P和Sn-CoOOH-P),并分别在其表面构建了OH吸附的结构。优化后的结构表明,两者均因相邻吸附位点间距较短而形成稳定的共吸附构型(分别为对称的Co-OH-Co与不对称的Co-OH-Sn)。吉布斯自由能计算表明,CoOOH-P的OH吸附能(-1.563 eV)低于Sn-CoOOH-P(-0.605 eV),说明前者对OH的吸附更强。进一步通过投影态密度分析发现,在对称吸附构型Co-OH-Co中,两个Co的3d轨道均与OH中O的2p轨道能级接近,产生强烈的重叠,导致OH中间体过度稳定化,不利于后续的反应步骤。而在不对称吸附构型Co-OH-Sn中,Co 3d与O 2p保持强相互作用,而Sn 5p与O 2p轨道因能级失配,重叠较弱。这种差异使得整体对OH的吸附能适中,更有利于析氧反应进行。因此,引入d¹⁰构型金属构建不对称吸附构型,为缓解活性位点被OH堵塞提供了理论可行的途径。
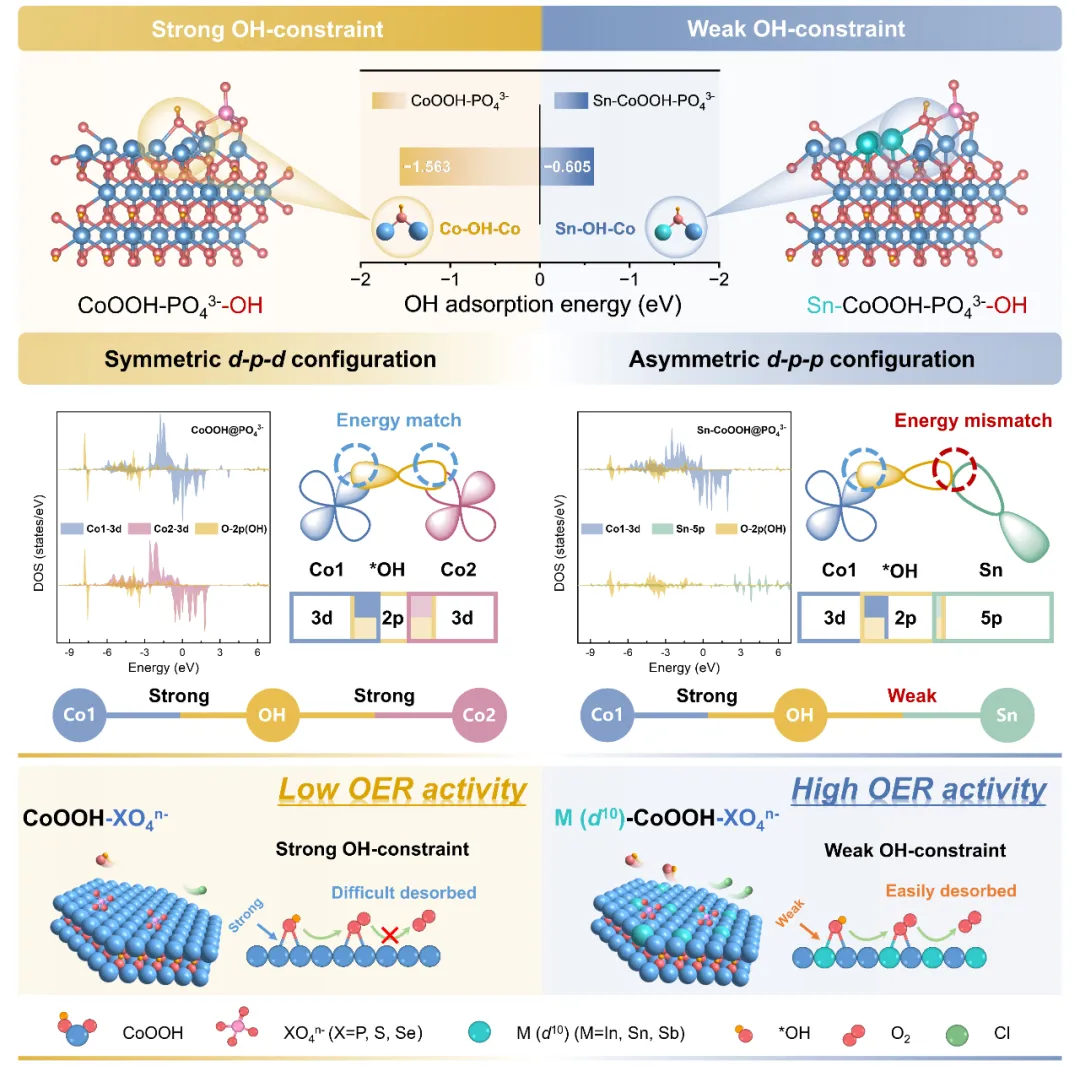
图1. 催化剂设计思路的可行性预测(图源:ACS Catal.)
(2)催化剂的合成与物理表征
基于上述对催化剂设计思路的理论预测,为了从实验上去验证该设计思路的可行性,本研究通过简单的水热、磷化及电化学氧化条件下驱动的原位重构三个步骤成功制备了Sn-CoOOH-P催化剂。通过对比重构前后催化剂的形貌变化(针状转变为片状)结合XPS与原位Raman分析证实了OER活性相CoOOH的形成。XAS图谱分析表明,Sn在Sn-CoP与Sn-CoOOH-P中的配位环境分别为Sn-P配位与Sn-O配位,且均未观察到对应于Sn-Sn金属键与Sn-O(SnO₂)金属键的特征峰,证明Sn在重构前后均是以替代部分Co位点的形式存在,这为不对称吸附构型Co-OH-M(d¹⁰)的形成提供了实验上的证据。
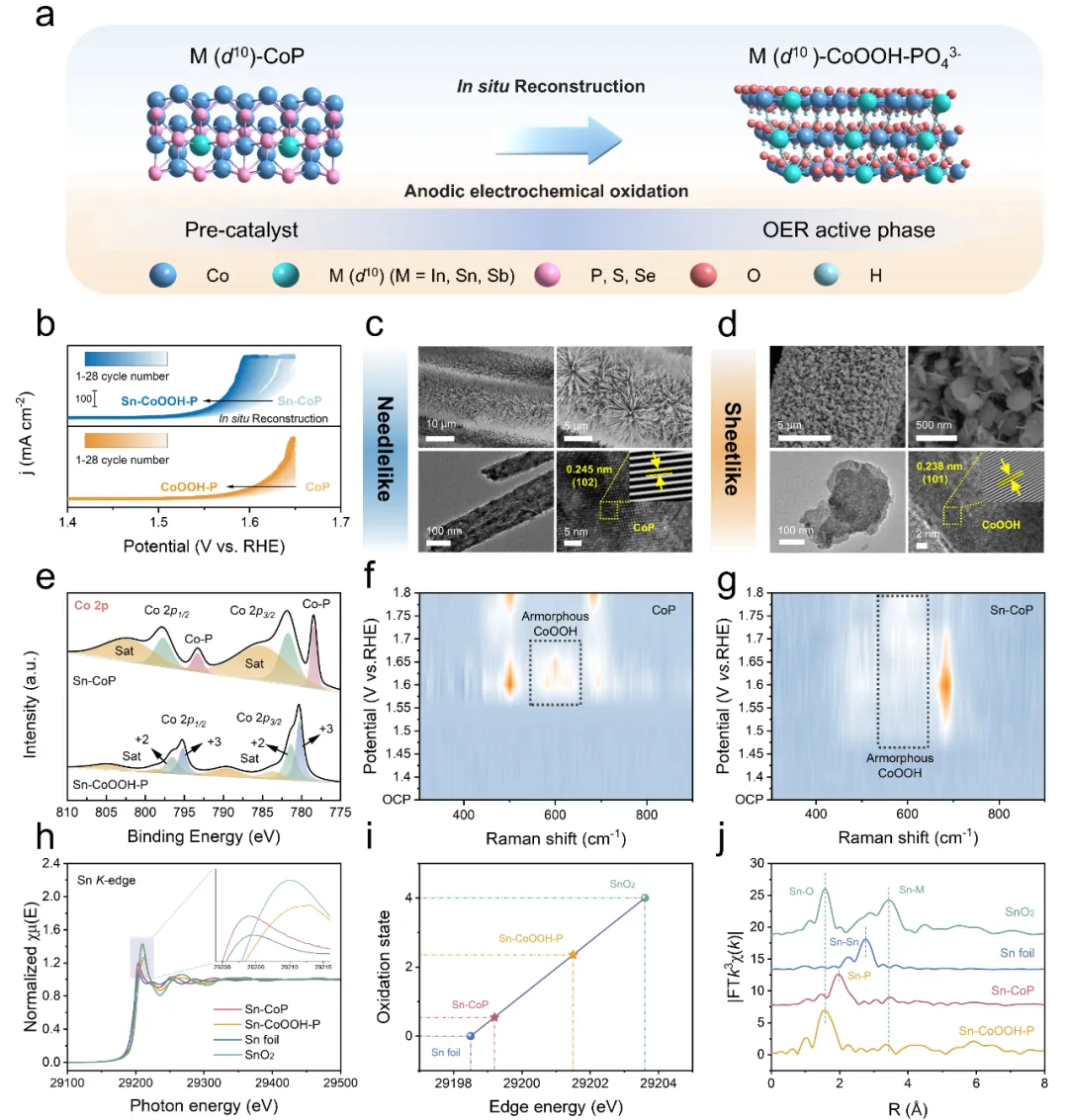
图2. 催化剂的形貌与物理表征(图源:ACS Catal.)
(3)电化学性能
随后采用标准的三电极体系对所设计的催化剂进行了一个初步的电化学性能探究。得益于d¹⁰构型Sn的引入,所设计的催化剂在模拟海水中表现出优异的OER活性,仅需264 mV与342 mV的过电为即可达到10 mA cm⁻²与100 mA cm⁻²的电流密度。更为重要的是,Sn-CoOOH-P在10 mA cm⁻²的电流密度下,在模拟海水中稳定运行55 h,展现出了出色的催化稳定性。
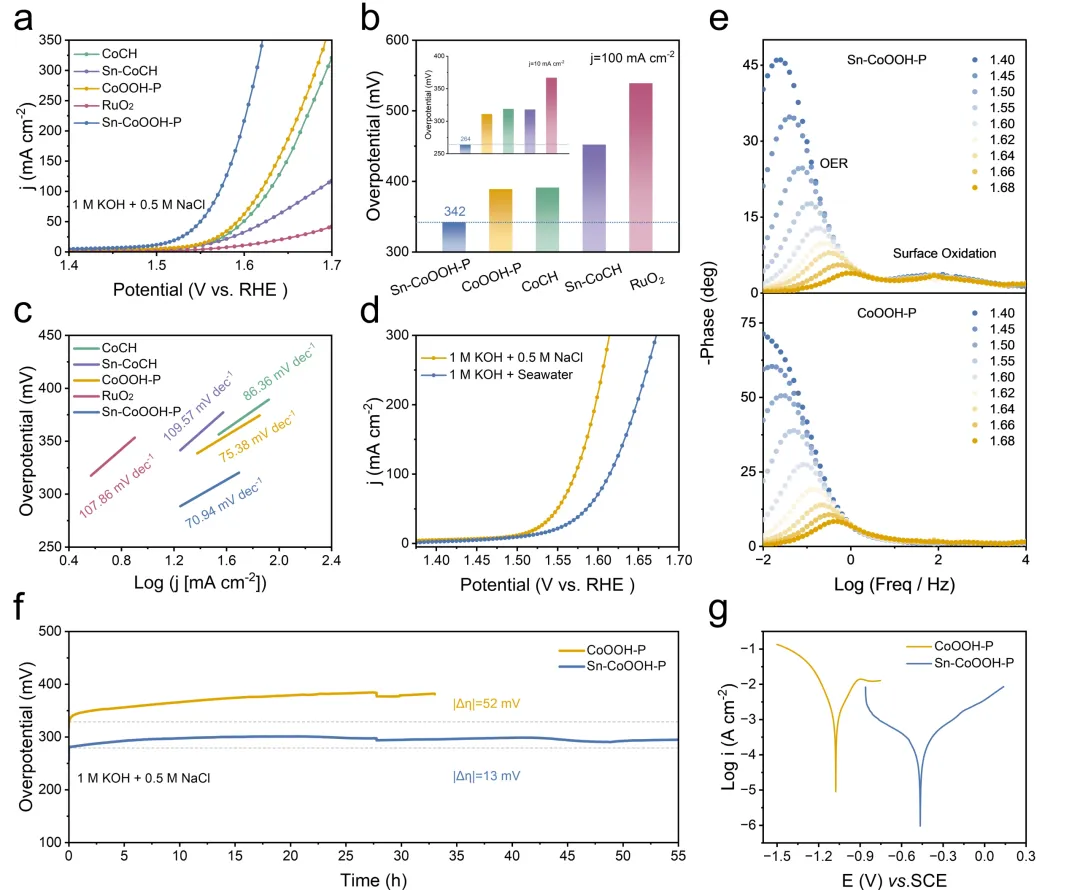
图3. 催化剂的电化学性能(图源:ACS Catal.)
(4)AEMWE器件性能评估
为进一步评估Sn-CoOOH-P作为阳极催化剂在大电流密度下的实际应用前景,作者将其集成到膜电极中并组装了碱性阴离子交换膜电解槽(AEM)。该电解槽在碱性模拟海水与碱性天然海水中分别仅需1.713 V与2.053 V的槽压即可达到500 mA cm⁻²的电流密度。更重要的是,该电解槽在模拟海水中于750 mA cm⁻²的电流密度下稳定运行500h,在碱性天然海水中以500 mAcm⁻²的电流密度稳定运行1000h,电压仅分别上升54 mV与46 mV,表现出优异的催化稳定性。此外,作者进一步计算了电解槽在不同电流密度下的能耗与电解效率。结果表明,即使在1 A cm⁻²的大电流密度下,组装的电解槽在模拟海水与碱性天然海水中的运行效率仍分别达到约67.5%与53.0%,制氢能耗仅为4.39与5.58 KWh Nm⁻³ H₂。经济性分析表明,每单位汽油加仑当量氢气的生产成本可低至0.86美元(最高1.26美元),低于美国能源部2026年目标(2.00美元)。
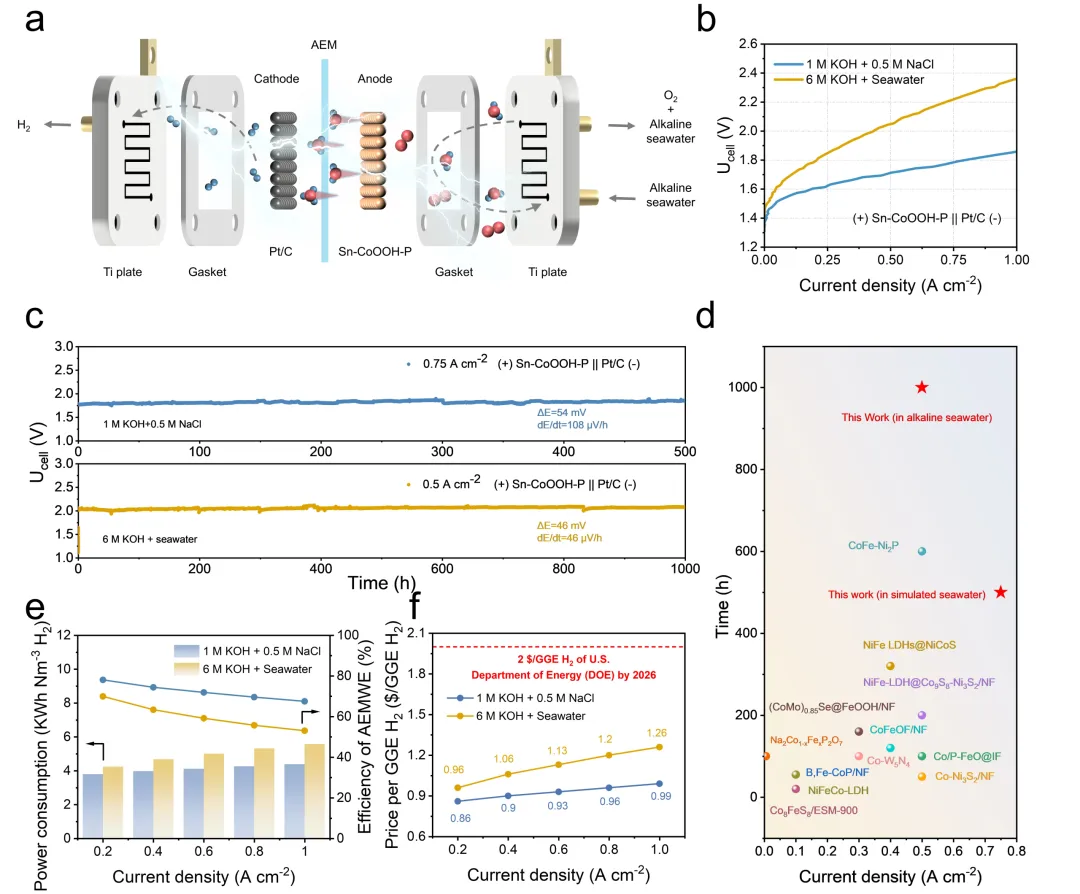
图4. AEMWE器件性能评估(图源:ACS Catal.)
(5)催化机制分析
为了阐明d¹⁰金属Sn调控OH的吸附及对海水电解OER活性影响的内在机制,作者进行了系统的DFT计算与原位表征分析。不同OH覆盖度下的吸附自由能计算表明,Sn的引入显著降低了OH的吸附强度,这归因于所形成的不对称吸附构型Co-OH-Sn有效缓解了OH的过度稳定化问题。原位红外结果表明,相较于CoOOH-P,Sn-CoOOH-P表面的含氧中间体覆盖度更低、特征峰开始出现的电位更高,证实了d¹⁰金属Sn掺杂有效削弱了含氧中间体的吸附,与计算结果相一致。基于AEM路径的吉布斯自由能计算表明,在CoOOH-P中,对称吸附构型Co-OH-Co对OOH产生强吸附,使O₂脱附成为速控步骤(能垒为1.68 eV)。而在Sn-CoOOH-P中,不对称吸附构型Co-OH-Sn通过较弱的d-p-p轨道相互作用缓解了对氧中间体的强吸附作用,将速控步骤转变为*O→*OOH,能垒降至0.93 eV,显著提升了OER反应动力学。
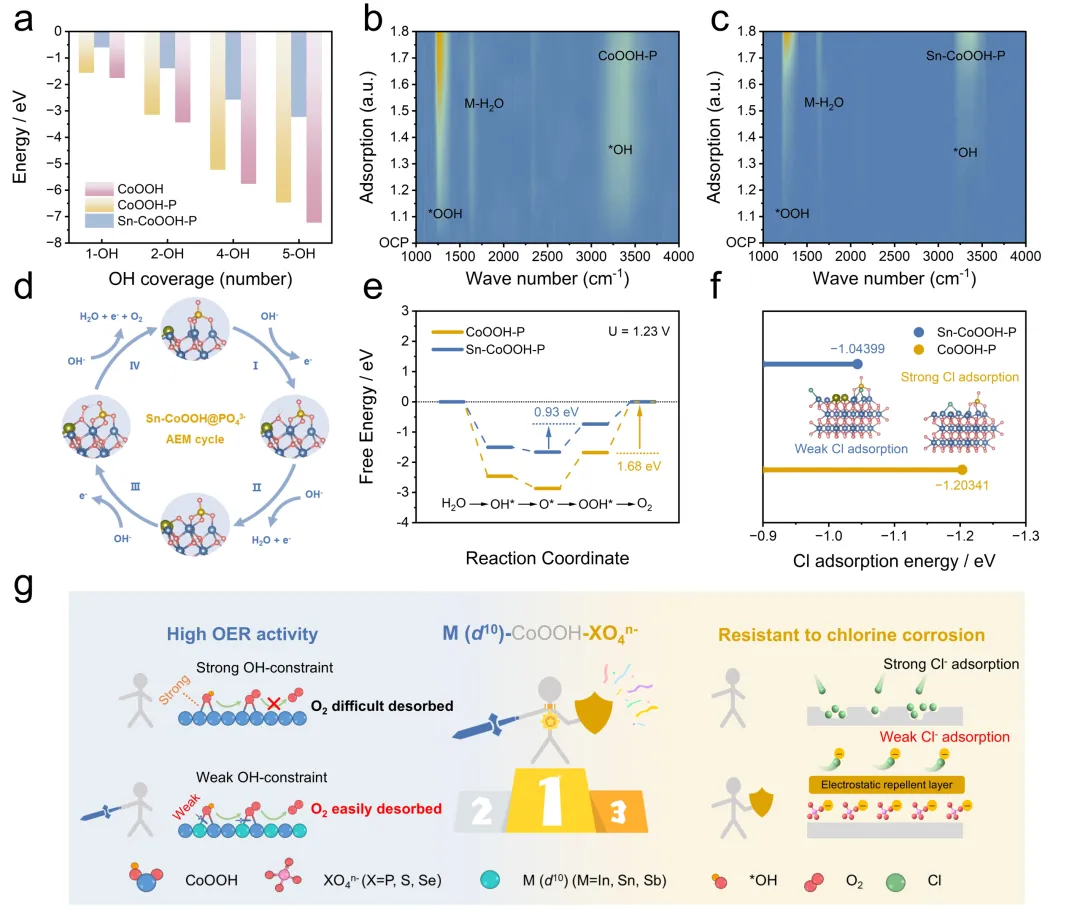
图5. 催化机制分析(图源:ACS Catal.)
总结
本文通过在Co-Xides(X=P、S、Se)中引入d¹⁰构型元素(In,Sn,Sb),利用电化学氧化条件下驱动的原位重构过程,成功构建了M(d¹⁰)-CoOOH-XO₄n-催化剂。理论计算与实验共同表明,所形成的不对称d-p-p构型(Co-OH-M(d¹⁰))能够有效削弱原有d-p-d构型(Co-OH-Co)对OH中间体的强束缚作用,从而缓解OH堵塞问题与促进活性位点的再生,显著提升了OER反应动力学。基于此策略制备的代表性催化剂Sn-CoOOH-P,在海水电解中表现出优异的催化活性和催化稳定性。本工作成功突破了传统催化剂在耐氯腐蚀性与高OER活性之间的权衡问题,为设计高效海水电解催化剂提供了新的思路与重要参考。

| 点击即可阅读合集 | ||
| 催化化学 | ||
| 分析化学 | ||
| 生物化学 |


