同一天,南京大学连发3篇Nature Chemistry!
- 2026-04-20 05:09:49

2026年3月3日,南京大学在化学研究顶级期刊 《Nature Chemistry》 同期发表3篇重磅研究论文。
①南京大学史壮志教授团队与合作者,通过光氧化还原催化实现了天然硫化砷矿物(如雌黄 As2S3)在可见光条件下与有机碘化物直接反应,一步构建结构多样化有机砷化合物,绕过有毒中间体,建立了一种安全、可持续且可放大的矿物到分子转化新范式。
②南京大学韩山雨、谢代钱教授团队与合作者,通过HOD光解离实验和全维量子计算,揭示了直接和间接解离路径在同一锥形交叉缝处产生的类似“单缝衍射”的动力学干涉,展示了单一反应路径内部产生的量子干涉。
③南京大学王国强团队联合湖南大学冯见君教授团队、中山大学钱宇、南京中医药大学张薇等团队,通过双环[1.1.0]丁烷与1,4-二噻烷-2,5-二醇的环加成反应,成功制备了2-硫杂双环[3.1.1]庚烷(thia-BCHeps),所得环加成产物可作为邻位取代苯和1,2,4-三取代苯的生物等排体,并通过晶体学分析、药代动力学比较及药物类似物的生物活性评价验证了其替代潜力。
01 通过光氧化还原催化实现矿物到分子的砷转移

文章链接:https://www.nature.com/articles/s41557-026-02064-2
有机砷化合物在医药和材料领域具有重要作用,但其合成长期依赖以As2O3(俗称砒霜)为起点的多步转化和有机金属亲核试剂(锂、镁、锌和铝基试剂),存在毒性高、步骤繁琐和官能团兼容性差等问题。 因此,发展一种绕过有毒前驱体、直接从天然砷矿构建As–C键的安全、高效且可持续的新方法,成为砷化学亟待解决的关键挑战。
对比了传统路径与本研究新路径,展示了新路径可以利用可见光驱动,通过使用易得的铱基光催化剂与叔胺联用,从雌黄(主要成分As2S3)一步构建 As-C 键。这一突破通过光激发的自由基路径,绕过了所有致命中间体,实现了无机砷直接转化为有机砷化合物。
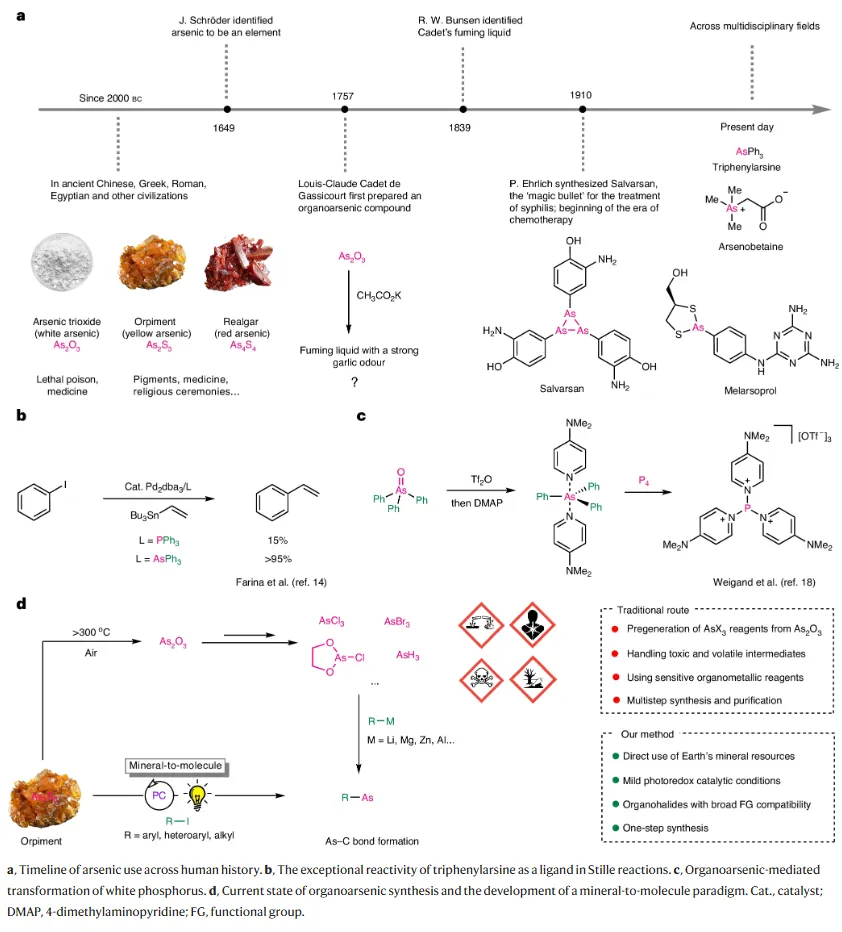
在可见光和铱光催化剂作用下,天然砷硫矿物(如雌黄)能够与碘代有机物发生光氧化还原反应生成有机砷化合物,其中光催化剂、光照、Et3N以及C–I键均为反应发生的关键条件。且该研究方案仅对反应时间敏感,对体系中的氧气水平、PhI 浓度、溶剂水分含量、温度波动和光照强度的变化保持了显著的稳定性。
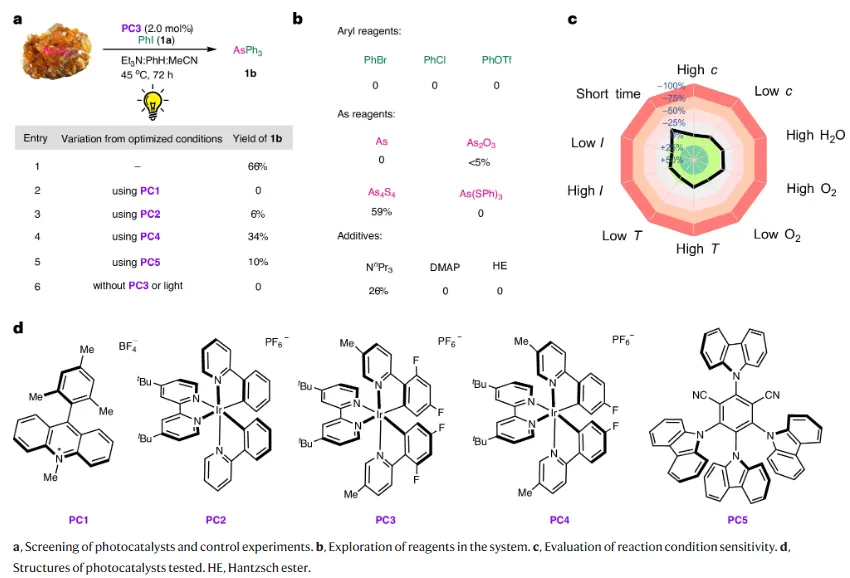
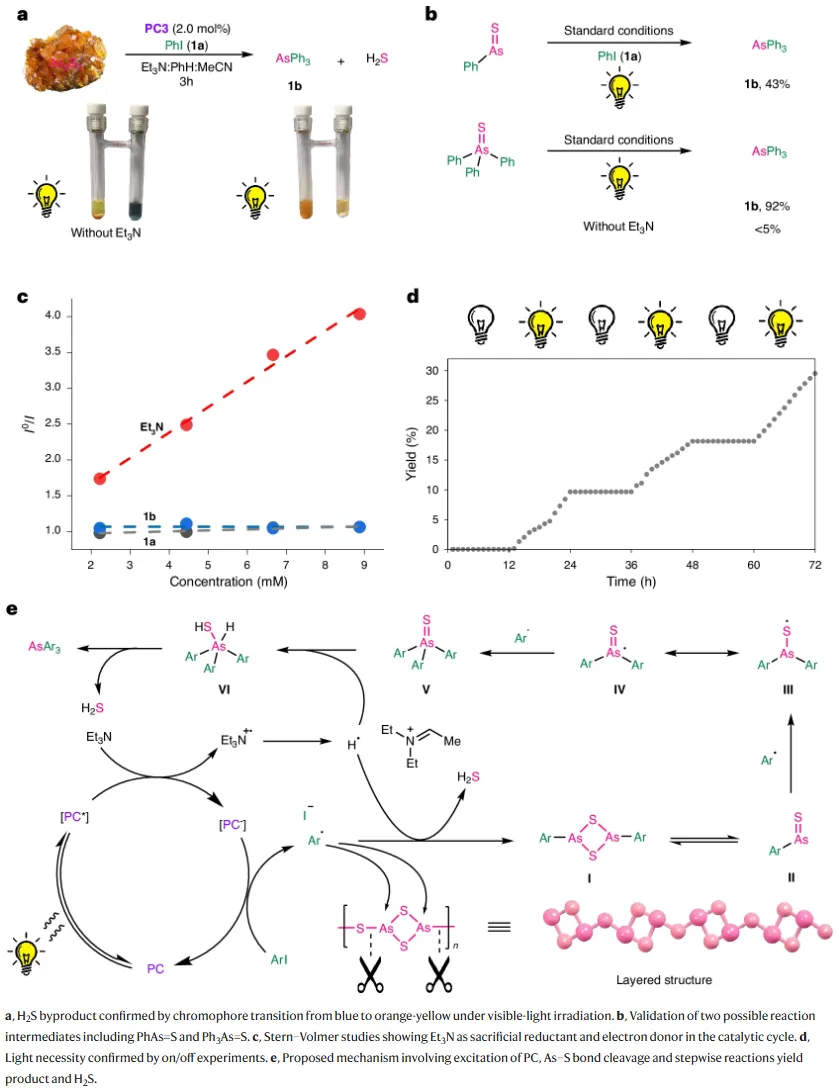
研究利用 H 型双室系统证明了,在光氧化还原体系中砷硫矿与有机碘化物反应会生成H2S副产物,且苯基硫化砷 (PhAs=S)等物种可能作为关键反应中间体参与反应过程。机理研究进一步证明Et3N优先猝灭激发态光催化剂PC3*并驱动电子转移生成芳基自由基(Ar•),这些自由基进攻砷中心,通过断裂延长的As–S键促进As–C键的形成,并最终生成有机砷化合物。
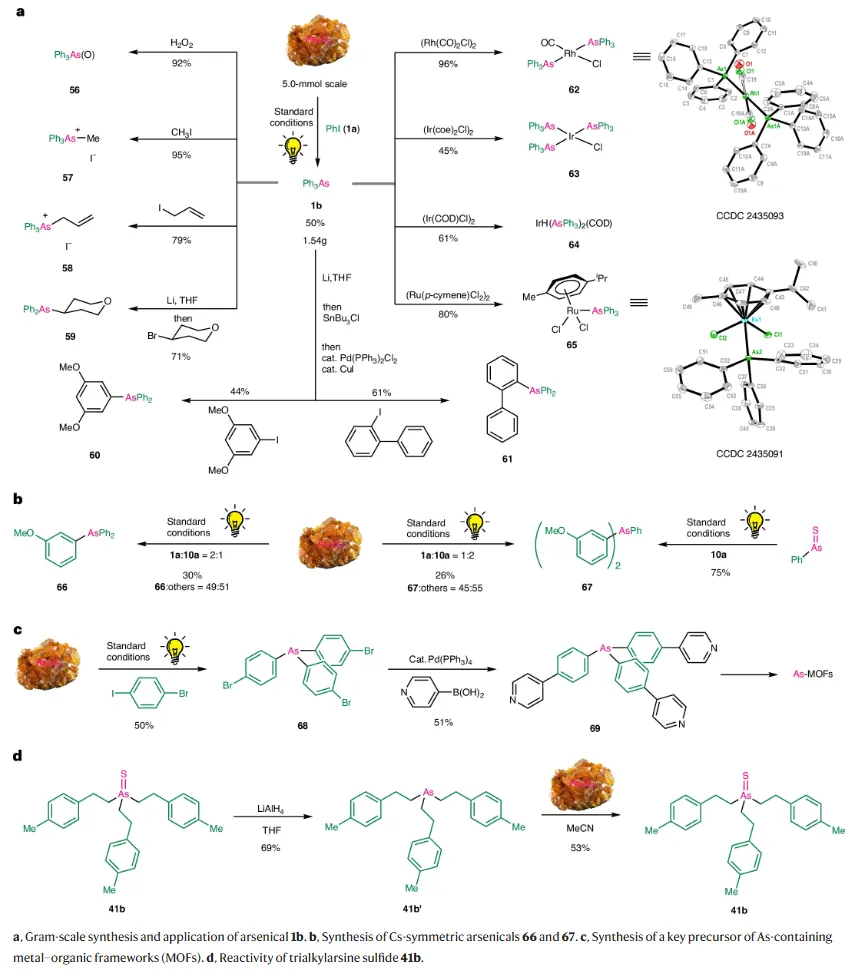
此外,该矿物到分子的策略具有良好的放大性和合成实用性,可克级制备有机砷化合物并进一步转化为氧化物、季铵盐、和多种金属配合物。该方法还提供了从雌黄直接制备含砷功能材料的更安全的替代路线,并揭示了三烷基砷与雌黄之间的可逆硫化反应。
02 HOD光解离过程中直接反应路径和间接反应路径之间的量子干涉

文章链接:https://www.nature.com/articles/s41557-026-02078-w
量子粒子的波动性可产生干涉效应,从而影响化学反应的结果。然而,过去观测到的干涉效应大多发生在两个空间上不同的反应路径上,类似于杨氏双缝实验。尽管量子干涉现象已在多种分子体系中被观察到,且锥形交叉在非绝热动力学中起关键作用,但通过锥形交叉的非绝热路径的内在动力学特征及其对可观测结果的微妙影响仍未完全阐明。
团队利用 Rydberg 标记飞行时间技术,测量了 HOD 在 121 nm 附近产物OD(X) 的转动分布。结果显示了其极其敏感的波长依赖性,波长仅微调约 0.1 nm,产物分布即发生剧变。
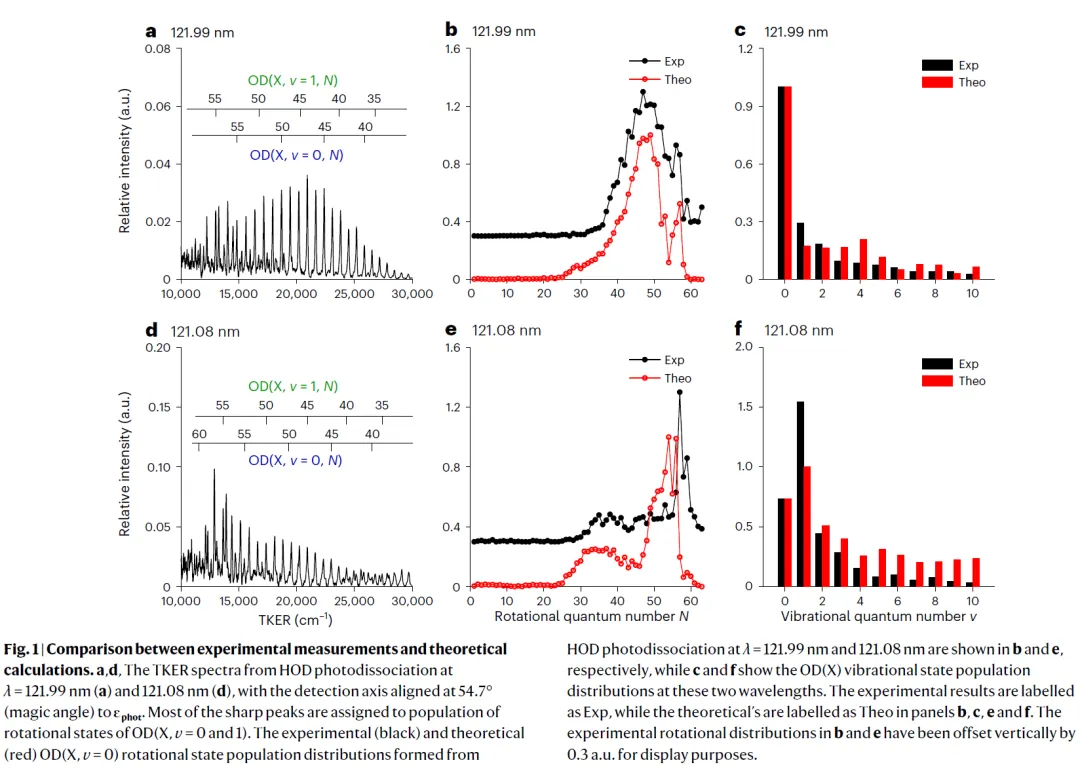
下图展示了锥形交叉处的路径分支。直接反应路径:分子在势能面的驱动下,以极快的速度直接穿过锥形交叉,随后解离,解离时间约为 9.5 fs;间接反应路径:分子并没有直接穿缝而过,而是在锥形交叉的上锥形区域发生瞬时俘获,随后再转移至基态,导致解离时间明显滞后,约为 46 fs。这种单一通道内的显著时间差转化为量子相位差,诱发类似“单缝衍射”的相消干涉。
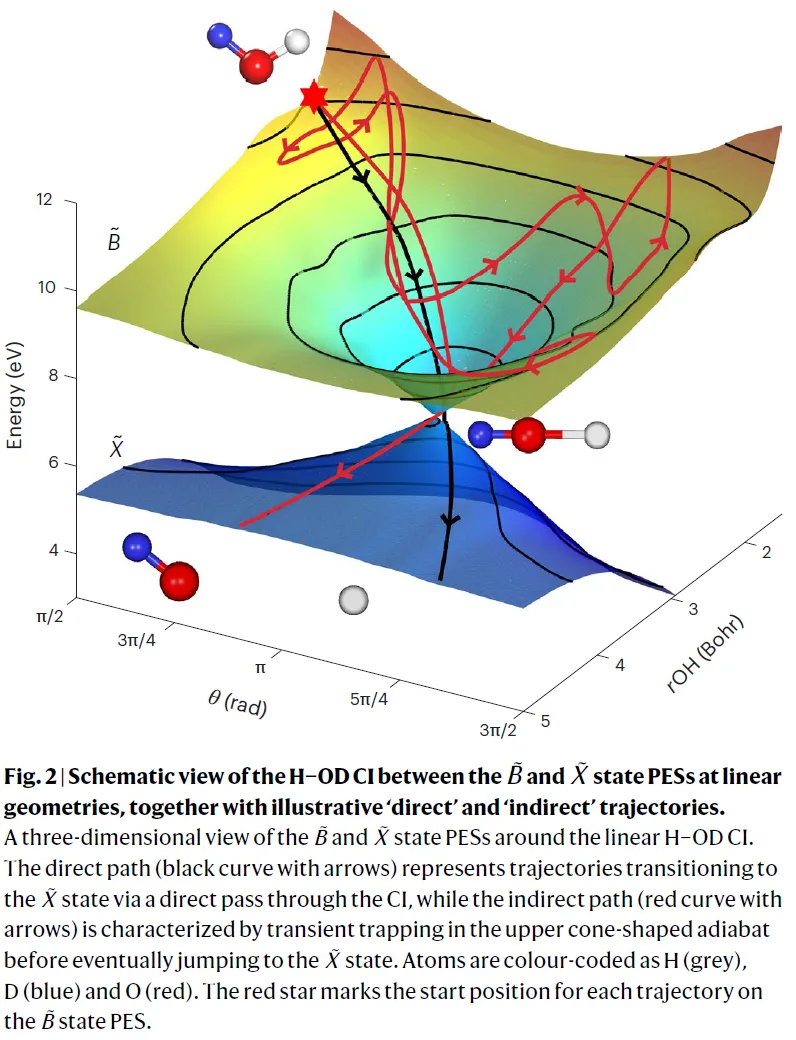
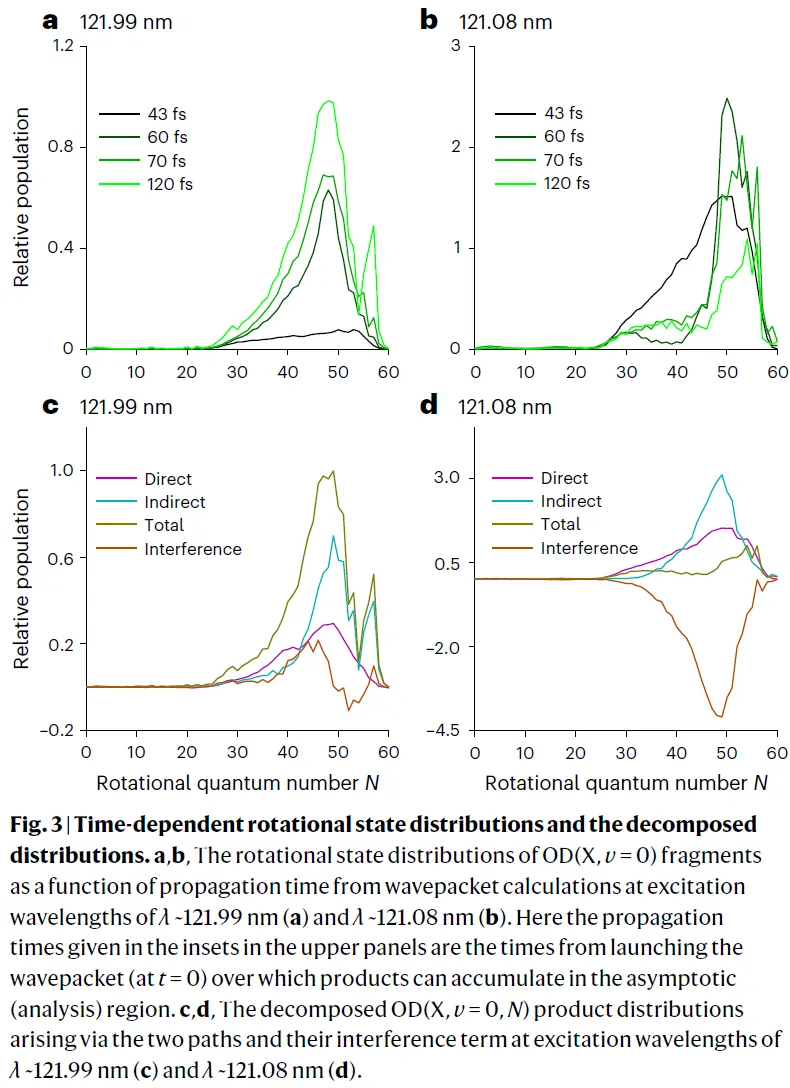
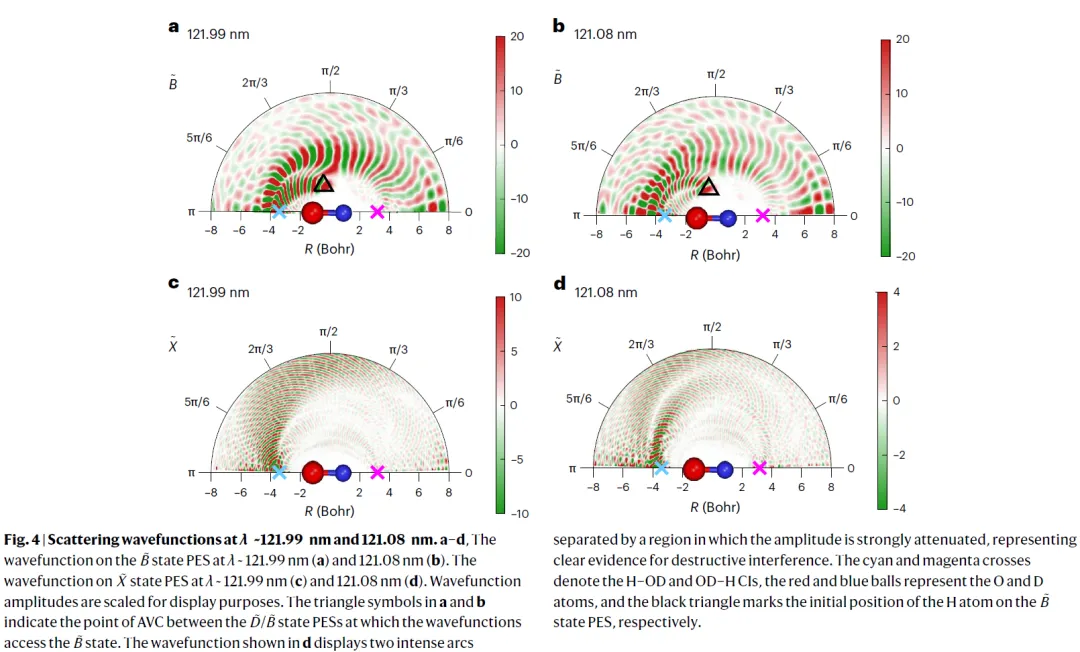
研究将总反应截面分解为直接项、间接项与干涉项,发现在 121.08 nm 波长下,干涉项在N≈25–40呈显著负值,导致该区间OD(X)分布抑制。在时间分辨的转动态分布中,产物初始积累后下降,进一步证实延迟贡献引起的破坏性干涉。此外,散射波函数分析显示,在121.08 nm波长下,X态波函数呈现双弧衰减区,提供了破坏性干涉的直接证据。而121.99 nm下表现为单弧,无明显干涉。研究还揭示了,改变波长会使干涉项在正负间翻转,并引起间接路径产物分布的振荡变化,从而显著影响总体OD(X)转动态分布,反映了非绝热动态对能量的敏感性。
03 由双环丁烷集体合成1,2,4-三取代、间位和邻位取代的芳烃生物等排体

文章链接:https://www.nature.com/articles/s41557-026-02097-7
在药物研发中,用富含C(sp3)杂化的生物等排体能改善药物的理化性质和药代动力学特征。然而,1,2,4-三取代苯环作为药物中常见的结构,其复杂的空间排布和难以合成的特性,特别是在催化(不对称)合成和验证多取代双环烃作为生物同分异构体时,仍然是药物化学中的一大挑战。
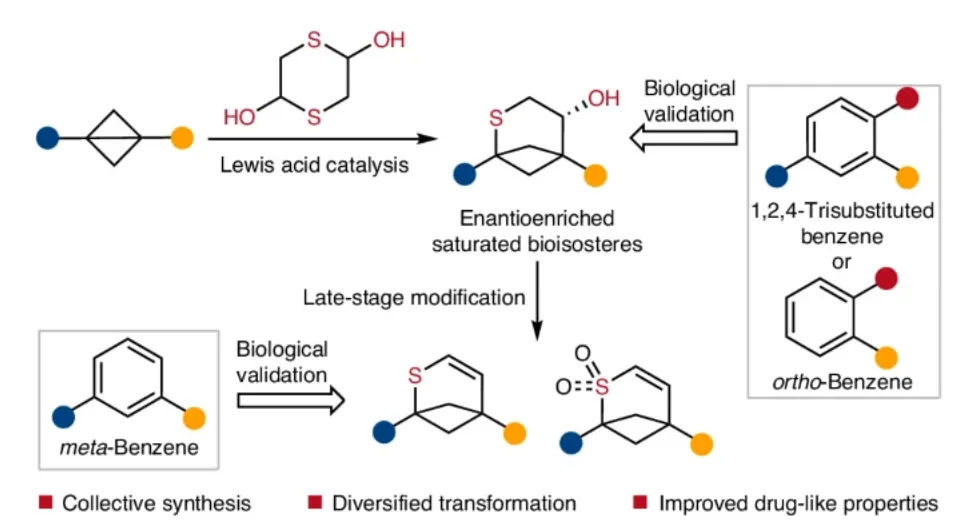
本研究开发了路易斯酸催化的双环丁烷与廉价巯基乙醛二聚体的[3+3]环加成反应,实现100%原子经济性和优秀的对映选择性,高效合成了具有两个和三个“出口向量”的2-硫杂双环[3.1.1]庚烷。通过系统性的后期官能团转化和硫原子氧化态调控,快速拓展了包括2-硫杂双环[3.1.1]庚烯在内的结构多样性化学空间,并可实现克级规模制备。基于5种苯环类药物活性分子的结构模拟,成功构建了12种双环[3.1.1]庚烷类三维类似物,显示了该方法在药物新骨架创制以及1,2,4-三取代、邻位和间位二取代苯环饱和生物电子等排体集群式合成方面的应用潜力。
出口矢量分析结合药代动力学、生物活性及安全性评价表明,上述硫杂双环[3.1.1]庚烷骨架作为苯环饱和生物电子等排体的可行性,且硫氧化态调控及等排体对映体对理化性质和生物活性的显著影响;此类硫杂双环[3.1.1]庚烷/烯衍生物作为可调节理化性质与生物活性的新型分子骨架,展现出广阔的药用前景,为药物发现的工具库增添了重要成员。
参考文献
1. Wang, Y., Ge, C., Glorius, F. et al. Mineral-to-molecule arsenic transfer via photoredox catalysis. Nat. Chem. (2026). https://doi.org/10.1038/s41557-026-02064-2
2. Wang, J., Luo, Z., Zhou, L. et al. Quantum interference between direct and indirect reaction paths in the photodissociation of HOD. Nat. Chem. (2026). https://doi.org/10.1038/s41557-026-02078-w
3. Wu, F., Wang, JJ., Xiao, Y. et al. Collective synthesis of 1,2,4-trisubstituted, meta- and ortho-substituted arene bioisosteres from bicyclobutanes. Nat. Chem. (2026). https://doi.org/10.1038/s41557-026-02097-7
版权声明
测试表征+计算+绘图

随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- 【幼儿食谱】宝贝私房菜-南京江南明珠幼儿园第一周幼儿食谱
- 软件研究院党委委员、副院长赵亚晖一行赴南京分院调研指导
- 【南京科普游】守护地球,“植”得“莓”好
- 兵工顶流・国防七子:南京理工大学报考全方位解读
- 我是甘肃人,去了一趟江苏省南京市,不吹不黑,南京市比网上评价的还要好!
- 活动预告|南京中医药大学2026年“学雷锋志愿服务月”启动仪式暨主题游园会
- 凝心聚力启新程 深耕教研促发展 ——南京市江北芳草园小学召开新学期期初教学工作会议
- 【招满截止】南京理工大学紫金学院丨人文与社会科学学院丨26年招聘专任教师招聘丨硕博丨人事代理
- 惊蛰春雷动,匠心启新程——南京烹饪技工学校2026年春季“开学第一课”燃情开讲
- 南京大学FinTech大模型实验室招募 | 斯坦福国际联培博士后(2026)
