
1、文章亮点
1、本文通过水热法在亲水碳布上生长Sn掺杂的钴羟基前驱体;随后通过低温磷化处理得到Sn–CoP;最后通过电化学原位重构形成Sn–CoOOH–PO43-活性相。该催化剂在模拟海水中达到10 mA cm-2和100 mA cm-2的过电位分别仅为264 mV和342 mV。在阴离子交换膜电解槽中,Sn–CoOOH–P在模拟海水中以750 mA cm-2稳定运行500小时,在天然海水中以500 mA cm-2稳定运行1000小时。氢气成本为0.86美元/kg,低于美国能源部2026年目标。
2、本文提出了一种非对称的d−p−p构型Co−OH−M(d10),以解决钴基化合物在OER过程中因OH中间体过度稳定而脱附困难的问题。作者将具有d10电子构型的金属引入钴基磷化物中,构建了非对称d-p-p电子构型。与传统对称的Co–OH–Co构型相比,d10金属的引入导致轨道能级失配,削弱了OH的吸附强度,从而降低表面OH覆盖度,提升析氧反应动力学。本文利用XPS证实Sn掺杂诱导Co缺电子态,同时原位拉曼光谱也表明Sn–CoP在更低电位下出现CoOOH特征峰,表明Sn加速了表面,促进重构形成CoOOH活性相;XANES和EXAFS证明Sn以取代Co位点形式存在,形成Sn–P–Co和Sn–O–Co结构;DFT计算表明非对称Co–OH–Sn构型通过弱d-p-p轨道相互作用削弱OH吸附,降低析氧反应能垒;原位ATR-FTIR证实Sn–CoOOH–P表面氧中间体覆盖度更低。2、全文速览
钴基磷化物、硫化物和硒化物(Co-Xides, X = P, S, Se)因其在海水电解中良好的耐腐蚀性而备受关注,这源于其重构形成的含氧阴离子层(PO43-, SO42-, SeO42-)能够通过静电作用排斥Cl-的吸附。然而,在重构后的CoOOH物种中,由于刚性双位点桥连构型(Co−OH−Co)的存在,OH物种被过度稳定,这显著提高了后续析氧反应步骤的能垒。为了克服析氧反应速率决定步骤的高能垒问题,引入具有d10构型的元素M(M = In, Sb, Sn),构建了一种非对称的d−p−p构型Co−OH−M(d10),该构型利用轨道能量和对称性的失配,打破了OH的过度稳定态,并削弱了OH吸附。原位表征和密度泛函理论计算证实,这种非对称的Co−OH−M(d10)吸附构型降低了表面OH覆盖度,从而显著提升了海水电解中析氧反应的活性。值得注意的是,代表性的Sn−CoOOH−PO43-催化剂表现出了优异的催化性能和耐久性:在模拟海水电解中,于750 mA cm-2(1.78 V)条件下实现了1000小时的长期稳定性;在碱性天然海水电解中,于500 mA cm-2(2.05 V)条件下实现了500小时的长期稳定性。
3、背景介绍
利用地球丰富的海水资源替代淡水,通过电解水制氢,对于可持续发展和氢经济至关重要。然而,碱性海水中存在的Cl-(约0.5 M)会导致阳极材料腐蚀并生成有毒的Cl2,严重损害海水电解的稳定性和安全性。近期研究表明,钴基磷化物、硫化物和硒化物被证明是用于海水电解的高效非贵金属阳极催化剂。其卓越的性能源于电化学氧化过程中的表面重构,生成了含氧阴离子修饰的CoOOH物种。这种富含带负电含氧阴离子的表面通过静电排斥作用抑制Cl-吸附,有效防止了Cl-的侵蚀,赋予了催化剂优异的抗氯腐蚀性能。然而,CoOOH的催化活性受限于含氧中间体(如OH、O、OOH)的过度稳定,这是由于相邻Co位点之间较短的原子间距以及中间体与两个Co中心之间的有效轨道重叠,导致活性位点被阻塞,并在后续析氧反应步骤中产生动力学限制。
通常,在对称的Co−OH−Co桥连构型中,由于能级接近,桥连O的2p轨道会同时与两个相邻Co位点的d轨道杂化,形成刚性的d−p−d电子结构。这种d−p−d耦合将电子转移限制在Co−OH−Co构型内,诱导了强烈的电子局域化,并通过增强的π电子反馈作用,在热力学上稳定了OH吸附,从而阻碍了随后的OH去质子化步骤。鉴于此,破坏d−p−d构型中强烈的电子局域化对于缓解OH的过度稳定至关重要。因此,提出用具有全充满d壳层的金属(d10金属)取代Co位点,以构建非对称的d−p−p构型。与d区金属的d轨道相比,p区的d10金属的p轨道在能级排列、轨道弥散程度等方面表现出更显著的差异,这导致了较弱的M(d10)−OH键,从而为吸附位点赋予了更大的灵活性,并促进了后续的析氧反应步骤。
基于上述考虑,本文考虑将一系列具有d10电子构型的元素M(如In、Sn和Sb)引入到磷化钴前驱体中。经过电化学重构过程,形成了M(d10)−CoOOH−PO43-活性相,从而构建出非对称的Co−OH−M(d10)吸附构型。由于M(d10)金属的p轨道与OH中O的2p轨道之间轨道重叠较少且能级失配,由此形成的M(d10)−OH键具有较弱且方向性较差的特征,与强吸附的对称Co−OH−Co结构相比,这削弱了Co−OH−M(d10)构型中整体的OH吸附强度。此外,这种非对称构型通过轨道能量失配也能有效削弱Cl⁻的吸附,从而同时提升了析氧反应活性和耐氯腐蚀性能。4、图文解析
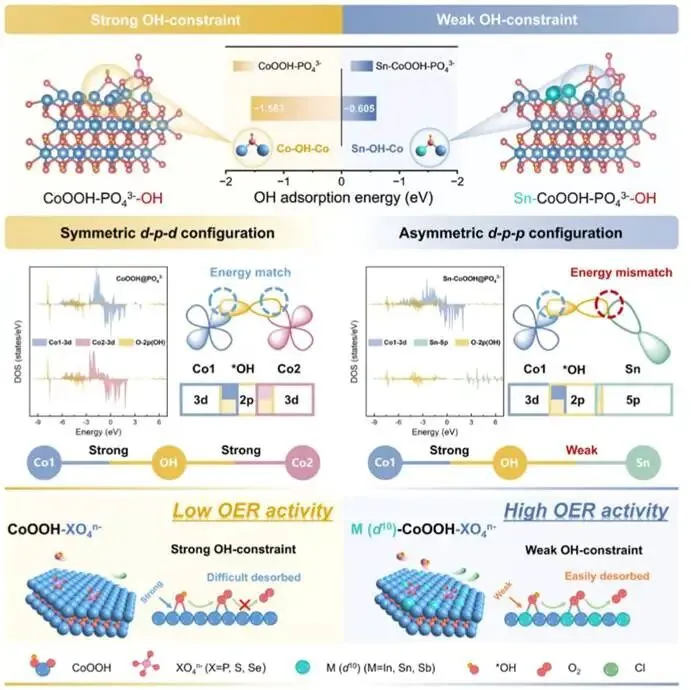
图1. 对称吸附结构Co−OH−Co与不对称吸附结构Co−OH−M(d10)对OH的吸附机理示意图。
首先进行密度泛函理论计算以预测上述设计概念的可行性。首先构建了PO43-修饰的CoOOH和PO43-修饰的Sn−CoOOH模型,其中选择Sn作为代表性的d10金属来阐明全充满d壳层对调控吸附强度的普遍效应。如图1所示,与Sn−CoOOH−P相比,CoOOH−P表现出更低的OH吸附吉布斯自由能,这表明OH在CoOOH−P上的吸附更强。为了更深入地理解控制吸附行为的潜在电子相互作用,分析了Sn−CoOOH−P和CoOOH−P上吸附OH的投影态密度。投影态密度分析表明,在对称的Co−OH−Co构型中,由于能级接近,每个Co原子的3d轨道与O的2p轨道表现出强烈的轨道重叠。然而,这种双位点吸附导致了OH中间体的过度稳定,这对后续的析氧反应步骤不利。相比之下,在非对称的Co−OH−Sn构型中,Co的3d轨道与O的2p轨道保持强相互作用,而Sn的5p轨道与O的2p轨道由于能级失配,其重叠显著减弱。这种差异化的相互作用导致了适中的整体吸附能,更有利于析氧反应过程。
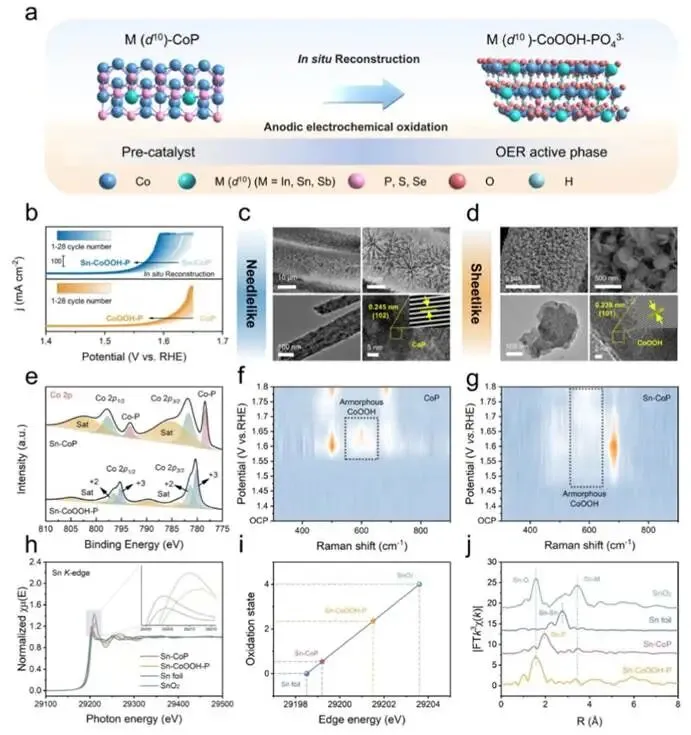 图2. (a) M(d10)−CoP在析氧反应过程中表面重构的示意图。(b) Sn−CoP和CoP在1.0 M KOH + 0.5 M NaCl中经过不同循环次数后的LSV极化曲线(经90% iR补偿)。(c, d) Sn−CoP和Sn−CoOOH−P的SEM和TEM图像。(e) Sn−CoP和Sn−CoOOH−P的Co 2p区域XPS精细谱。(f, g) CoP和Sn−CoP的原位拉曼光谱。(h) Sn箔、SnO2、Sn−CoP和Sn−CoOOH−P的归一化Sn K边XANES谱。(i) Sn−CoP、Sn−CoOOH−P、Sn箔和SnO2的Sn K边吸收能量与氧化态的关系。(j) 相应的k3加权傅里叶变换谱。
图2. (a) M(d10)−CoP在析氧反应过程中表面重构的示意图。(b) Sn−CoP和CoP在1.0 M KOH + 0.5 M NaCl中经过不同循环次数后的LSV极化曲线(经90% iR补偿)。(c, d) Sn−CoP和Sn−CoOOH−P的SEM和TEM图像。(e) Sn−CoP和Sn−CoOOH−P的Co 2p区域XPS精细谱。(f, g) CoP和Sn−CoP的原位拉曼光谱。(h) Sn箔、SnO2、Sn−CoP和Sn−CoOOH−P的归一化Sn K边XANES谱。(i) Sn−CoP、Sn−CoOOH−P、Sn箔和SnO2的Sn K边吸收能量与氧化态的关系。(j) 相应的k3加权傅里叶变换谱。
图2b所示,Sn−CoOOH−P催化剂的合成步骤包括水热过程、磷化处理以及原位表面重构。如图2b所示,两种样品在本征析氧反应活性上均表现出逐渐增强的趋势,表明在连续线性扫描伏安循环过程中发生了表面氧化并逐步形成了重构相。扫描电子显微镜图像显示,经过连续线性扫描伏安测试后,催化剂的形貌从最初的纳米针演变为均匀排列的纳米片。随后,通过X射线粉末衍射、高分辨透射电子显微镜和能量色散X射线光谱元素分布图进一步评估了详细的结构信息,证实所得的纳米片结构对应于CoOOH。
在Sn−CoP和Sn−CoOOH−P的Co 2p谱图中,与Sn−CoP相比,Sn−CoOOH−P中对应于Co−P键的峰消失,同时在780.2 eV和795.4 eV处出现了对应于高价Co3+的新特征峰,表明形成了新的CoOOH相。在Sn−CoOOH−P的P 2p谱图中,所有与M−P键相关的峰均已消失,仅剩下对应于P−O键的峰。表明CoP晶格中的所有磷原子均以PO43-离子的形式浸出并吸附在催化剂表面。采用原位拉曼光谱监测析氧反应过程中催化剂表面状态的演变。如图2f、2g所示,CoP和Sn−CoP均在526 cm-1和691 cm-1处表现出典型的Co−O特征信号。位于570−630 cm-1的宽峰归属于CoOOH相的Eg对称性的Co−OH振动模式。与CoP相比,Sn−CoP催化剂在更低的外加电位下就出现了对应于CoOOH相的特征峰,表明Sn调控后表面重构过程加快。
测量Sn−CoP和Sn−CoOOH−P的Sn K边X射线吸收近边结构谱。如图2h所示,Sn−CoP和Sn−CoOOH−P的阈值E0值介于Sn箔和SnO2之间,表明这四个样品中从1s到外壳层轨道的跃迁能量顺序为:Sn箔 < Sn−CoP < Sn−CoOOH−P < SnO2。通过E0的函数关系进一步评估了Sn−CoP和Sn−CoOOH−P中Sn的氧化态,表明Sn−CoP和Sn−CoOOH−P中Sn的平均氧化态分别为+0.54和+2.35。如图2j所示,Sn K边FT-EXAFS谱在1.56 Å处显示出一个特征峰,该峰归属于Sn−O键。未出现与SnO2中Sn−Sn键相关的峰,表明在电化学重构过程中Sn并非以金属Sn和SnO2的形式存在。相反,它通过部分取代CoOOH晶格中的Co位点存在,形成Sn−O−Co结构。
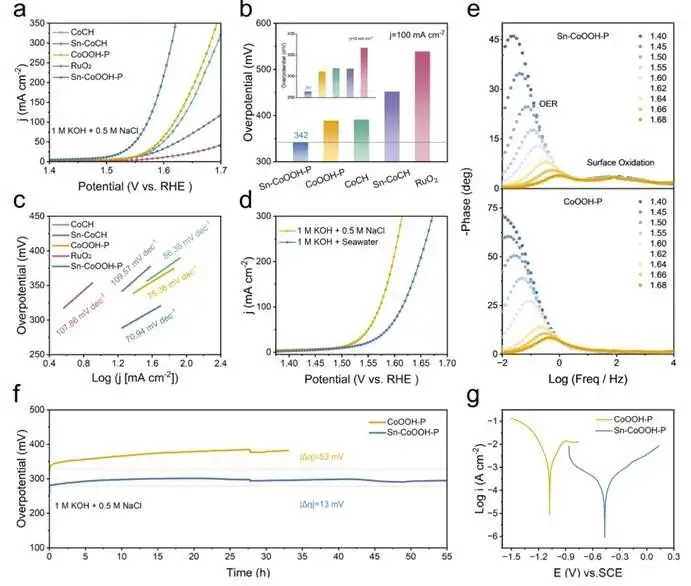
图3. 电催化析氧反应。(a) 不同催化剂在1.0 M KOH + 0.5 M NaCl中的极化曲线。(b) 各样品在1.0 M KOH + 0.5 M NaCl中达到10和100 mA cm-2电流密度所需的过电位。(c) 相应的塔菲尔曲线。(d) Sn−CoOOH−P电极在不同电解液中测试的极化曲线。(e) CoOOH−P和Sn−CoOOH−P的波特相位图。(f) CoOOH−P和Sn−CoOOH−P电极在1.0 M KOH + 0.5 M NaCl中,10 mA cm-2恒电流密度下的长期稳定性测试。(g) Sn−CoOOH−P和CoOOH−P电极在天然海水中的腐蚀极化曲线。
在模拟海水(1 M KOH + 0.5 M NaCl)中,采用标准三电极体系评估了Sn−CoOOH−P的析氧反应活性。如图3a所示,Sn−CoOOH−P展现出优异的析氧反应活性,在模拟海水中达到10和100 mA cm-2的电流密度仅分别需要264 mV和342 mV的过电位。这一性能不仅优于CoOOH−P、Sn−CoCH、CoCH和空白碳布,也超过了商业RuO2。Sn−CoOOH−P的塔菲尔斜率为70.94 mV dec-1,明显低于CoOOH−P、CoCH、Sn−CoCH和RuO2。为了模拟相关应用环境,进一步研究了Sn−CoOOH−P在1 M KOH和海水体系中的析氧反应活性。如图3d所示,Sn−CoOOH−P在1 M KOH+海水中的性能与在1 M KOH+0.5 M NaCl中相比出现一定程度的下降,这是由于天然海水污染物导致电极中毒。Sn−CoOOH−P催化剂达到10和100 mA cm-2电流密度仍仅需277 mV和385 mV的过电位。通过在不同电位下的原位电化学阻抗谱分析,优化的Sn−CoOOH−P在低频区域表现出较小的相位峰角度,表明Sn的掺入增强了电荷转移过程和析氧反应动力学。
为了评估催化剂的耐久性,在碱性模拟海水中采用三电极体系进行了计时电位测试。恒流测试55小时后,Sn−CoOOH−P达到10 mA cm-2所需的过电位仅增加了13 mV。相比之下,CoOOH−P仅经过35小时的耐久性测试就增加了52 mV,突显了Sn−CoOOH−P优异的长期耐久性。值得注意的是,经过55小时的耐久性测试后,Sn−CoOOH的片状形态保持完好,没有任何明显的聚集或脱附现象。为了更深入地了解Sn−CoOOH−P的耐腐蚀性,在不添加导电KOH的纯天然海水中评估了相应的腐蚀极化曲线。与原始CoOOH−P催化剂相比,Sn−CoOOH−P表现出更高的腐蚀电位和更低的腐蚀电流密度,证明Sn掺杂同时增强了催化剂的耐氯腐蚀性能。
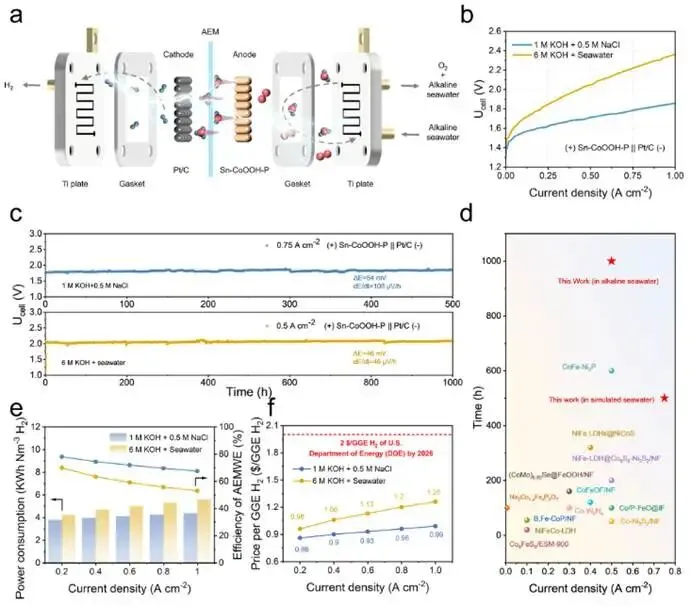
图4. (a) 阴离子交换膜电解槽示意图。(b) 阴离子交换膜电解槽的极化曲线。(c) Sn−CoOOH−P||Pt/C在模拟海水和天然海水电解槽中,于750 mA cm-2和500 mA cm-2条件下的稳定性测试。(d) Sn−CoOOH−P在阴离子交换膜电解槽中的电流密度和耐久性与文献报道的其他电催化剂的比较。(e)采用Sn−CoOOH−P||Pt/C的阴离子交换膜电解槽的能耗和效率。(f) 该阴离子交换膜电解槽生产1.0 kg氢气的预估成本。
为了评估Sn−CoOOH−P作为阳极催化剂的实用性,将其集成到膜电极组件中,组装成碱性阴离子交换膜水电解槽(图4a)。如图4b所示,该AEMWE器件在500 mA cm-2的电流密度下,于碱性模拟海水和碱性天然海水中分别实现了1.713 V和2.053 V的全电池电压。更重要的是,该电解槽在模拟海水中于750 mA cm-2条件下稳定运行500小时,在碱性天然海水中于500 mA cm-2条件下稳定运行1000小时,电压增幅分别仅为54 mV和46 mV(图4c)。与近期报道的其他钴基催化剂相比,Sn−CoOOH−P展现出了可观的性能和耐久性。随后,进一步计算了AEM电解槽在不同电流密度下的能耗和效率,以评估其应用可行性。如图4e所示,即使在高电流密度1 A cm-2下,在模拟海水和碱性天然海水中仍能分别保持约67.5%和53.0%的运行效率,能耗分别仅为4.39和5.58 kWh N m-3 H2。作为体现AEM电解槽经济性的关键因素,根据图4f的计算,制氢的价格低至0.86美元,这低于美国能源部设定的2026年2.00美元的目标。
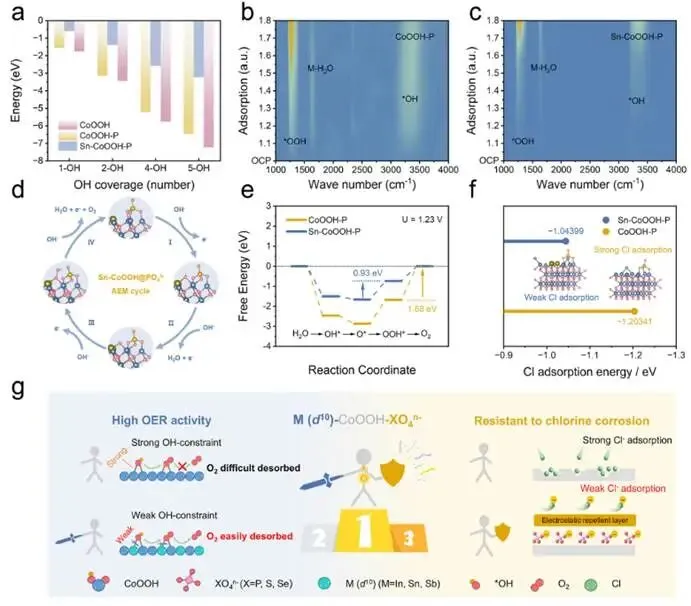
图5. (a) CoOOH、CoOOH−P和Sn−CoOOH−P在不同OH覆盖度下的计算自由能。(b, c) CoOOH−P和Sn−CoOOH−P的原位ATR-FTIR光谱。(d) Sn−CoOOH−P的析氧反应机理示意图。(e) 基于吸附质析出机制计算的CoOOH−P和Sn−CoOOH−P的Co位点上析氧反应自由能图。(f) CoOOH−P和Sn−CoOOH−P的Cl吸附能密度泛函理论计算。(g) Sn−CoOOH−P上O2脱附增强和耐氯腐蚀能力提升的示意图。
首先计算OH在不同覆盖率下在原始CoOOH、PO43-修饰的CoOOH和PO43-修饰的Sn−CoOOH上的吸附自由能。如图5a所示,原始CoOOH在所有覆盖率下均表现出最强的OH吸附能力,这体现在其最负的吸附自由能上。PO43-的引入通过对接近的OH-产生静电排斥作用来削弱OH吸附,从而形成了抑制OH吸附的能垒。此外,Sn的掺入使得OH吸附强度更显著地降低。结论表明,由于非对称Co−OH−Sn吸附构型的形成,Sn的掺入有效降低了OH覆盖度,从而解决了OH吸附过强的问题,并促进了后续的析氧反应步骤。进一步采用原位傅里叶变换红外光谱监测析氧反应过程中催化剂表面关键氧中间体的动态演变。如图5b、5c所示,CoOOH−P和Sn−CoOOH−P的原位反射强度热图谱显示了析氧反应的三个关键反应中间体。析氧反应过程中OOH信号的出现表明两种催化剂均遵循吸附质析出机制进行反应,这为密度泛函理论计算提供了可靠依据。此外,与Sn−CoOOH−P相比,在CoOOH−P上,这些物种的特征峰出现在更低电位,并且随施加电位升高其强度增加更为显著。上述对比表明Sn−CoOOH−P上的含氧中间体覆盖度较低,证实了d10金属Sn的掺入削弱了这些中间体的吸附。
计算析氧反应过程中各基元步骤的吉布斯自由能。对于原始CoOOH−P,对称吸附构型Co−OH−Co对吸附的OH施加了强结合效应,这导致沿着吸附质析出机制路径的含氧中间体结合过强。这种强限域作用特别稳定了OOH物种,使得从OOH释放O2在能量上最为困难。因此,速率决定步骤为OOH → O2,计算得到的能垒为1.68 eV。然而,当引入具有d10构型的Sn形成非对称吸附构型Co−OH−Sn时,OH的吸附强度被削弱。结果,这种被削弱的OH吸附传递给了下游中间体,使其具有适中的结合强度,尤其降低了OOH脱附/析氧的能量损失。因此,OOH → O2步骤不再是自由能变化最大的步骤,速率决定步骤转变为O → *OOH,其最大ΔG减小为0.93 eV,这与过电位降低相一致。此外,与原始CoOOH−P相比,Sn−CoOOH−P对初始OH吸附步骤表现出更高的能垒,从而降低了后续步骤的整体能垒,促进了析氧反应动力学。此外,考虑到海水体系中Cl-的存在,理论上进一步研究了材料表面的Cl吸附。与CoOOH−P相比,Sn−CoOOH−P表现出更高的Cl吸附能垒,这归因于Sn与Cl之间的轨道能量失配,表明Sn−CoOOH−P在海水电解液中不易受Cl⁻干扰,并具有优异的耐氯腐蚀性能。5、总结与展望
综上所述,针对钴基化合物固有的局限性,本文提出了一种将d10构型金属原子掺入钴基化合物,以构建非对称d−p−p构型用于海水电解的策略。实验和理论计算均证明了非对称d−p−p构型在削弱d−p−d构型对OH的强限域效应中的关键作用,这有利于*OO脱附,从而增强析氧反应动力学。因此,代表性的Sn−CoOOH−P催化剂在海水电解中表现出优异的催化活性和耐久性。本工作克服了耐氯腐蚀性与高析氧反应活性之间传统的权衡关系,为合理设计高效海水电解催化剂提供了宝贵思路。
6、文章链接
Alleviating OH Blockage: Asymmetric Adsorption Configuration of Co–OH–M (d10) for Seawater Electrolysis
https://pubs.acs.org/doi/10.1021/acscatal.5c08465


如需转载或投稿请联系我们:energy_catalysis@163.com
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
