南京师范大学杨维本教授团队AFM:以废治废-废塑料综合利用控制1O2提升废水处理效率.
第一作者:崔力
通讯作者:杨维本
通讯单位:南京师范大学
论文DOI:10.1002/adfm.75143
逻辑链条: 广泛背景(PMS高级氧化技术)→ 核心问题(¹O₂选择性低且催化剂回收困难)→ 现有方法局限(传统MOF粉末难回收、活性位点单一)→ 提出本文核心概念(混合自旋轨道工程)和策略(废PET制备MOF并原位生长于Kevlar纤维)→ 阐述假设的机制链条(混合配体 → 高/低自旋Co(II)共存 → 协同活化PMS → 选择性生成¹O₂)→ 预告研究成果(实现高效、稳定、可回收的水污染治理)。
在复杂废水中实现高PMS利用效率和对单线态氧(1O2)的可控选择性活化仍是一大挑战。论文构建了一种废弃聚对苯二甲酸乙二醇酯(PET)衍生的MOF基催化体系(W-Co-MOF@Kevlar),用于PMS吸附活化和1O2生成,实现持续、近乎完全的污染物去除。复合催化剂显著促进1O2的演化(金属原子利用率(MAUR)为1.6-1.8×105 mg CIP mol metal-1,是仅用LS-Co(Co-MOF)的50倍)。在PET衍生的异质结构中,自旋轨道工程诱导了Co自旋轨道能级分裂,从而实现电子群体从低自旋态向高自旋态的部分转移,由此产生的非简并轨道将能级向上移动,激活额外的三维轨道电子态。这种电子结构优化重新导向了PMS的激活途径,有利于高效的1O2生成。此外,利用废弃PET作为原料,并通过共价键固定于纤维基材,使W-Co-MOF@Kevlar催化剂具有更高的环境可持续性,并具有显著的工业实用潜力。
单线态氧(1O2)因能实现化学选择性氧化、具有未被占据的π*轨道,且对复杂的水相基质具有很强的耐受性,因此正越来越多地被应用于废水净化和选择性转化。由于PMS的活化过程常与产生SO4•-和•OH的自由基途径发生竞争,这可能降低1O2的选择性并增加副产物生成的风险。因此,实现高效的PMS利用及可控的1O2选择性活化,仍是PMS驱动型AOP面临的核心挑战。
我们再看这篇文章的DFT理论计算部分,作者团队的操作堪称教科书级的“理论指导实践,再反哺机理”,远非简单的“算算而已”!整个计算逻辑清晰地揭示了“混合自旋轨道工程”如何精准调控PMS(过氧单硫酸盐)的活化路径,直指单线态氧(¹O₂)的选择性生成。
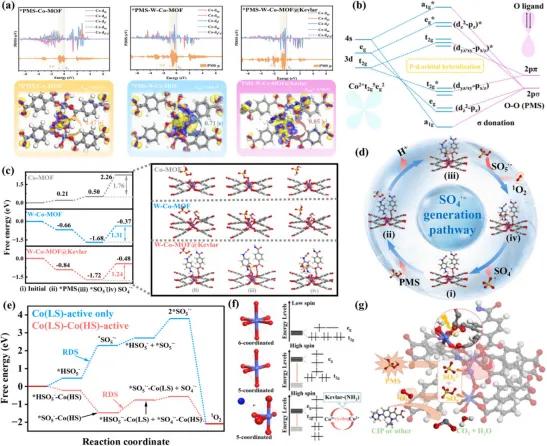
1. 直击核心——轨道杂化与电荷转移(图4a, 4b):
作者没有泛泛而谈,而是通过计算态密度(PDOS)和电荷密度差分,精准地回答了“为什么高自旋(HS)Co位点比低自旋(LS)Co位点活性更高?”这个核心问题。
PDOS(图4a, 4b) 一针见血地指出:在高自旋(HS)的W-Co-MOF@Kevlar体系中,Co的d轨道(特别是d₂², dₓz, dᵧz)与吸附的\PMS中间体中氧原子的p轨道在费米能级附近发生了强烈的杂化和自旋劈裂。这种“轨道匹配”带来了显著的“σ接受-π反馈”协同作用,有效削弱了\PMS的O-O键。相反,在低自旋(LS)的Co-MOF中,轨道能量不匹配,杂化很弱,O-O键几乎不受影响。
电荷密度差分(图4a) 则将这种轨道相互作用“可视化”。图中清晰显示,电子从HS-Co位点(W-Co-MOF@Kevlar)大量转移给了\PMS(0.85 |e|),导致其吸附能极强(-1.34 eV)。而LS-Co位点(Co-MOF)的电子转移和吸附能则弱得多。这就从根源上解释了HS-Co位点是启动PMS活化的“发动机”。
2. 量化反应路径,锁定速控步(图4c, 4e):
理论计算最强大的地方在于能“看清”反应的每一步。作者通过计算反应势能面,精准锁定了整个催化循环的瓶颈。
\SO₄⁻的生成(图4c, 4d): 计算表明,在HS-Co位点上,O-O键断裂生成\SO₄⁻的能垒(RDS)仅为1.24 eV。而在LS-Co为主的Co-MOF上,这个能垒高达1.76 eV,几乎无法逾越。这直接从动力学上证明了HS-Co是活化PMS的关键。
¹O₂的选择性生成(图4e): 这是最精彩的部分!作者对比了两种可能的¹O₂生成路径:LS位点上两个\SO₅⁻的“自偶联”,以及HS位点生成的\SO₄⁻与LS位点上\SO₅⁻的“交叉偶联”。计算结果明确显示,“交叉偶联”的能垒远低于“自偶联”,揭示了HS-Co和LS-Co位点之间存在一种“分工协作”的协同机制。
3. 构建完整机理闭环(图4f, 4g):
最后,作者将所有计算结果整合成一个清晰、自洽的物理图像。
自旋调控电子构型(图4f): 通过配体工程(引入MHET)诱导Co从LS态部分转变为HS态,抬高了d带中心,优化了轨道杂化,这是催化活性提升的“电子根源”。
Kevlar纤维的作用(图4f): Kevlar上的给电子基团(-NH₂)为Co活性中心提供了“电子补给”,稳定了催化循环,这是材料高稳定性的“结构保障”。
HS-LS协同机制(图4g): HS-Co位点负责“暴力”破解PMS的O-O键,生成高活性的\SO₄⁻;而LS-Co位点则温和地稳定\SO₅⁻中间体。两者协同作用,高效、选择性地生成了¹O₂。
总而言之,这篇论文的DFT计算部分不是对实验结果的简单附会,而是一场深刻的理论剖析。它从“自旋态改变”这一量子现象出发,通过“轨道杂化”这一桥梁,完美解释了“HS-LS位点协同催化¹O₂选择性生成”这一复杂的宏观催化现象。整个论证过程逻辑严密、层层递进,充分展现了理论计算在揭示复杂催化机理中的强大力量。
摘要是文章的“精华浓缩版”,咱们按照“背景-问题-方案-亮点-意义”的框架来解析:
研究背景: 过氧单硫酸盐(PMS)是一种用于废水处理的多功能氧化剂,其中,选择性地将其活化为单线态氧(¹O₂)是一种高效且抗干扰能力强的策略。
存在的挑战/问题: 在复杂的实际废水中,要实现PMS的高利用率,并能可控地、高选择性地生成¹O₂,这仍然是一个巨大的挑战。传统的催化剂往往会同时产生硫酸根自由基(SO₄•⁻)和羟基自由基(•OH),降低了选择性。
解决方案: 作者提出了一种“变废为宝”的策略。他们用废弃的PET塑料瓶作为原料,制备了一种新型钴基金属有机框架(W-Co-MOF),并将其原位生长在Kevlar(凯夫拉)纤维上,构建了一种MOF-纤维异质结构(W-Co-MOF@Kevlar)。
核心亮点与机理:
1. 性能优异: 该催化剂能持续、高效地去除污染物(如环丙沙星CIP),金属原子利用率(MAUR)比传统Co-MOF高出50倍,且在真实水体中抗干扰能力强。
2. 机理创新: 通过“自旋轨道工程”(spin-orbital engineering),在PET衍生的异质结构中诱导了Co的自旋轨道能级分裂。这促使部分电子从低自旋(LS)态跃迁到高自旋(HS)态。
3. 路径重定向: 这种优化的电子结构改变了PMS的活化路径,极大地促进了高选择性¹O₂的生成,而非自由基的产生。
应用意义: 该研究不仅提出了一种高效的水处理催化剂,更重要的是,它利用废弃PET作为原料,并将其稳定地固定在纤维上,大大增强了材料的环境可持续性和工业化应用的潜力。
前言部分,是作者引导读者进入其研究领域的“路径图”:
开篇点题,指出过一硫酸盐(PMS)作为一种多功能氧化剂在废水处理中的应用前景。然后直指核心——在复杂废水中,同时实现高PMS利用率和可控、高选择性地生成单线态氧(¹O₂)仍然是一个巨大挑战。接着阐述研究¹O₂选择性的难点:催化剂表面通常是异质的,多种活性位点共存,导致反应路径复杂,难以将¹O₂的选择性归因于单一设计的位点。结论自然导出:需要一个结构明确、可系统调控的平台来研究结构与选择性之间的关系。
话锋一转,作者介绍了金属有机框架(MOFs)作为理想模型平台的优势:晶体结构明确、配位环境可编程。但紧接着就点出其应用的“痛点”——粉末形态难以回收、在水中可能不稳定、存在金属浸出风险。这直接引出了将MOFs固定在稳定载体上的必要性。随后,作者介绍了凯夫拉(Kevlar)纤维作为载体的优越性:高强度、耐化学腐蚀,并且其表面可通过酸化处理引入活性基团,为MOF的原位生长和化学键合提供了可能。
作者终于亮出了自己的创意!“To address these challenges, we develop a waste-derived, structurally defined Co-MOF-on-fiber platform…”。他们明确提出,要利用废弃的PET塑料作为原料,通过一种创新的“晶体水一锅法”合成一种混合配体Co-MOF(W-Co-MOF),并将其原位、共价键合地生长在凯夫拉纤维上。作者还特意提到,这种混合配体结构能稳定相邻的高自旋(HS)和低自旋(LS)Co(II)位点。最后,作者清晰地阐述了他们的核心论点:HS-Co(II)和LS-Co(II)位点通过自旋协同作用,前者负责PMS的初步活化,后者稳定关键中间体并决定反应选择性,从而高效地将PMS活化路径引导至生成¹O₂。这不仅实现了“以废治废”,还解决了MOF催化剂的实际应用难题。
MHET共价桥联: MHET将W-Co-MOF与酸化Kevlar稳定耦合,驱动MOF在纤维表面共形、致密原位生长。
混合自旋工程:配体/应变协同重构Co晶场,稳定HS/LS Co(II)邻位点,定向促进PMS选择性生成1O2。
高MAUR与强抗干扰:HS/LS协同使CIP降解MAUR达1.6-1.8 × 105 mg mol−1(约50倍于LS),并具优异耐受性。
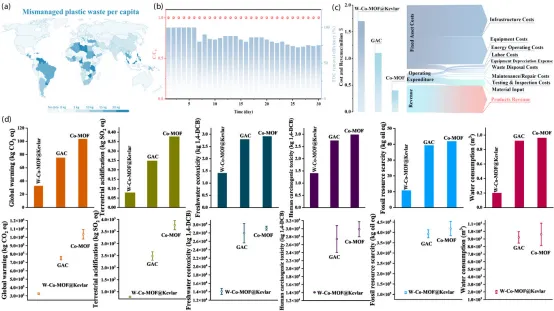
环境可持续性:废弃PET和Kevlar的协同驱动下,W-Co-MOF@Kevlar兼具“以废治废”的材料来源优势和较低的环境负荷;LCA和TEA显示其环境影响和经济竞争力优于Co-MOF和GAC。
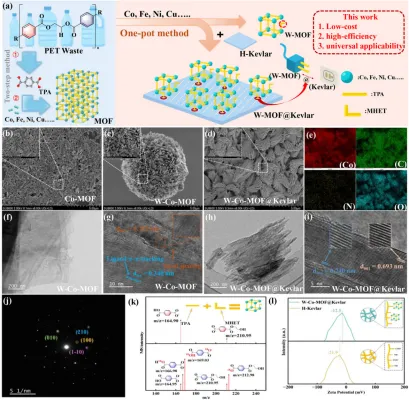
结构构筑与界面锚定:图1展示了由传统两步法Co-MOF到晶水驱动一锅法W-Co-MOF,再到W-Co-MOF@Kevlar的构筑过程与形貌演变;一锅法产物呈花状超薄纳米片组装结构,MHET桥联促使MOF在酸化Kevlar表面原位共形生长,TEM/SAED/元素映射及18O示踪共同证实了混配体参与、结构有序且界面稳定的共价锚定MOF-on-fiber异质结构成功构建。
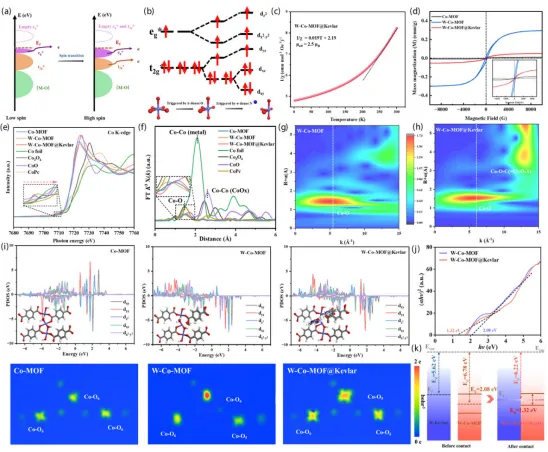
自旋态与电子结构调控: 图2系统揭示了W-Co-MOF@Kevlar中Co位点由单一LS向HS/LS共存状态的演变:磁学测试表明其存在混合自旋Co(II),XANES/EXAFS/WT-EXAFS证实Kevlar界面诱导的低配位畸变与Co-O/N重构,PDOS与能带分析进一步表明界面耦合可缩窄带隙、建立内建电场并提升费米能级附近电子态密度,从而为PMS活化提供更优电子结构基础。
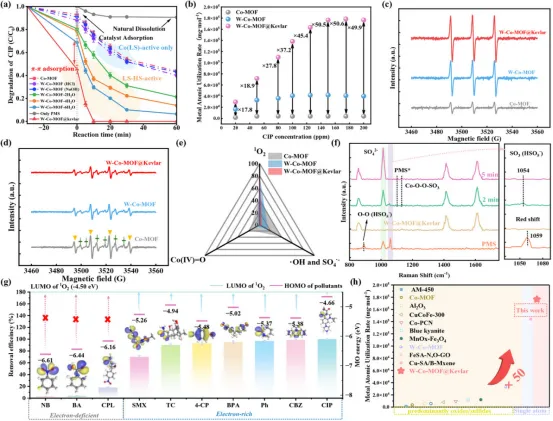
催化性能与1O2主导氧化:图3表明HS/LS-Co协同的W-Co-MOF@Kevlar在PMS活化降解CIP中表现出显著优于LS-only Co-MOF的活性和金属原子利用效率,MAUR达(1.6-1.8) × 105 mg CIP mol metal−1;EPR、猝灭实验、DPA探针和原位Raman共同证明该体系将PMS活化路径由•OH/SO4•−主导转向以1O2为主的非自由基路径,并对富电子污染物表现出更强选择性和优异矩阵耐受性。
自旋协同PMS活化机理:图4通过DFT计算阐明了HS/LS Co位点的分工协同机制:HS-Co因d轨道分裂与O-2p轨道耦合增强,更有利于PMS吸附、电子转移和O-O键断裂生成SO4•−相关中间体,而LS-Co则更倾向于稳定*SO5•−;两者跨位点协同降低了1O2生成能垒,从而实现低能垒、定向的1O2选择性形成。
工程适配与环境可持续性:图5展示了该材料从废PET/Kevlar资源化到实际水处理应用的工程潜力:W-Co-MOF@Kevlar在复杂水体、宽pH范围和连续流条件下保持高去除率、低Co浸出与长期结构稳定,同时TEA显示其经济性优于Co-MOF和GAC,LCA进一步表明其在18项环境影响指标中整体负荷最低,体现出突出的“以废治废”可持续优势。
研究结果表明,混合配体工程构筑的W-Co-MOF@Kevlar可在真实水体中实现对PMS活化路径的有效调控,并显著增强1O2生成选择性。其根源在于混合配体框架与纤维诱导界面效应共同重构了Co位点的局域晶场,稳定了相邻的HS/LS Co(II)活性中心,并赋予其自旋协同分工特征,从而调控PMS衍生关键中间体的演化过程。由此,PMS可经由双位点协同路径被选择性活化,并使氧化过程偏向以1O2为主导的非自由基机制;相比自由基主导路径,该机制通常更不易受到复杂基质干扰。得益于此,W-Co-MOF@Kevlar在连续流条件下、宽pH范围(3–11)内均可实现持续、高效的污染物去除,同时保持极低的Co浸出和极高的金属原子利用效率,从而降低二次污染风险与材料消耗。进一步的TEA和LCA结果表明,该体系相较传统Co-MOF和GAC具有更优的经济性与环境可持续性,显示出良好的实际应用前景。更重要的是,本研究将废弃PET来源配体化学与纤维支撑体上的界面自旋态调控相结合,为构筑高稳定性、高选择性的氧化还原催化材料提供了可推广的设计思路,也为废弃资源高值利用与绿色水处理材料开发提供了新的启示。
L.Cui, S.Liu, Y.Ge, et al. “Controlling 1O2 Evolution by Mixed-Spin-Orbital Engineering With a Waste-PET-Derived-MOF on Kevlar for Enhanced Water Decontamination.” Advanced Functional Materials (2026): e75143. https://doi.org/10.1002/adfm.75143课题组网站链接:http://hky.njnu.edu.cn/info/1191/12120.htm

 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
