机制创新:首次系统阐明 ZFP36/E2F1/ATF4 轴在 OS 中的调控作用,明确 ZFP36 通过靶向降解 E2F1,抑制下游 ATF4 转录,从而促进铁死亡、抑制 OS 恶性进展;
临床转化价值:ZFP36 在 OS 中显著低表达,可作为 OS 预后标志物及潜在治疗靶点,为 OS 精准治疗提供新方向;
多维度验证:从细胞、分子、动物多层面验证调控机制,同时结合临床样本分析,结论严谨可靠;
聚焦铁死亡与线粒体:深入解析 ZFP36 对铁死亡及线粒体功能障碍的调控,为 OS 铁死亡靶向治疗提供理论基础。
骨肉瘤(OS)好发于青少年,易转移、预后差,5 年生存率低,亟需阐明其分子机制。铁死亡参与肿瘤进展,ZFP36 作为铁死亡调控基因在 OS 中低表达,但其功能尚不明确。E2F1、ATF4 分别在肿瘤发生及铁死亡中发挥重要作用。本研究旨在探究 ZFP36/E2F1/ATF4 轴通过调控铁死亡及线粒体功能影响骨肉瘤进展的机制,为治疗提供新靶点。
如果你也想用同款思路分析,想学习如何复刻这类高分研究的同学,快关注后台咨询吧!生信解码站,助力每一个科研梦想!
如果你也想进行生信分析,快联系生信解码站,你的下一篇顶刊论文,就从这里开始啦!
骨肉瘤(OS)恶性程度高、预后差,亟需明确分子机制。铁死亡是 OS 进展关键,ZFP36 作为铁死亡调控基因在 OS 中低表达,但其功能及机制未明。E2F1、ATF4 分别参与肿瘤发生及铁死亡调控。本研究探究 ZFP36/E2F1/ATF4 轴对 OS 铁死亡及恶性进展的调控,为治疗提供新靶点。
临床与细胞验证:通过 GEO 数据集、Western blot、免疫组化,验证 ZFP36 在 OS 组织及细胞系中的低表达;
功能实验:在 MG63/U2OS 细胞中过表达 / 敲低 ZFP36,检测细胞增殖、克隆形成、迁移、侵袭能力,以及铁死亡相关指标(ROS、MDA、GSH、Fe²⁺、GPX4、SLC7A11)和线粒体功能;
机制研究:通过双荧光素酶报告基因、ChIP 等实验,明确 ZFP36 靶向 E2F1 3'UTR 促进其降解,E2F1 转录激活 ATF4 的调控轴;
体内验证:构建裸鼠异种移植瘤模型,验证 ZFP36 过表达对 OS 体内生长的抑制作用。
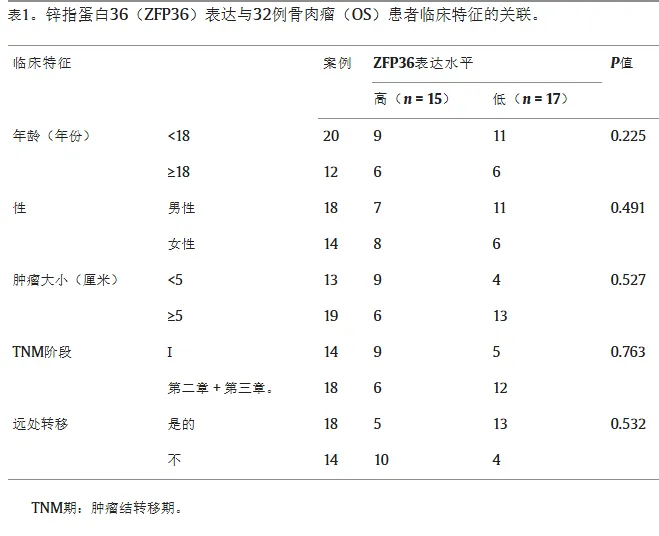
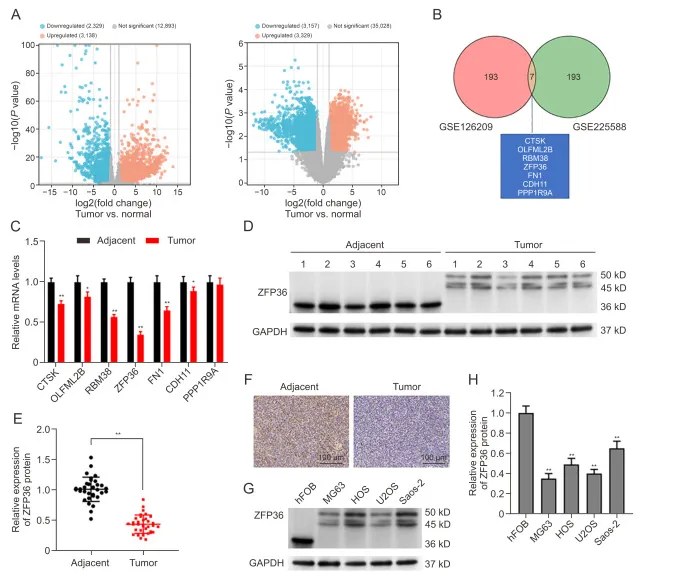
在OS患者和细胞中ZFP36下调
在两个 GEO 数据集中筛选出 7 个差异基因,其中 ZFP36 在骨肉瘤组织中 mRNA 水平下调最显著。Western blot 及 IHC 证实,ZFP36 蛋白在骨肉瘤组织及细胞系中均低表达,条带约 45–50 kD 并存在磷酸化修饰,提示 ZFP36 可能参与骨肉瘤进展。
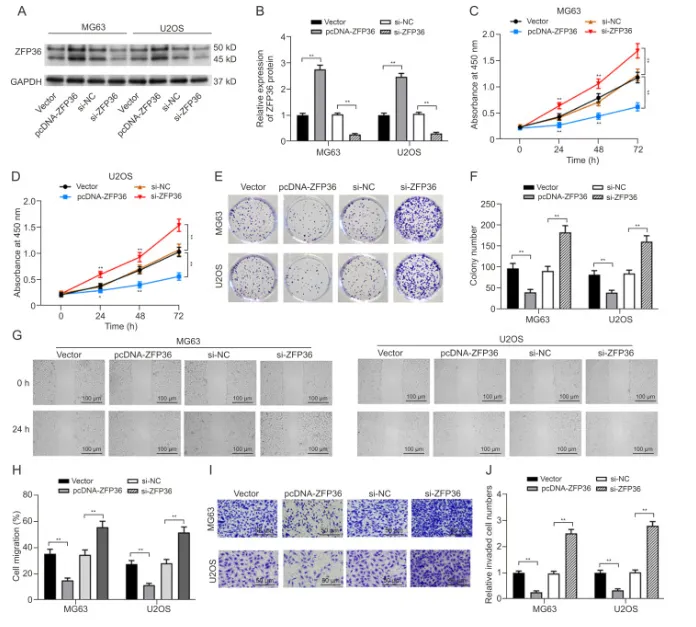
ZFP36过表达抑制了OS细胞的增殖、迁移和入侵
在 MG63 和 U2OS 细胞中验证 ZFP36 功能:过表达 ZFP36 可显著提高其蛋白水平,而 si-ZFP36 则有效敲低 ZFP36 表达。功能实验显示,ZFP36 过表达明显抑制骨肉瘤细胞的增殖、克隆形成、迁移及侵袭能力,敲低则呈现相反效应。
对比正常组织,膀胱癌中 ACTG1、C19orf57 显著上调,GRIK2、DYM、PTGER3 显著下调,ANK3、AK9 无明显差异。生存分析显示 ACTG1 等五个基因高表达与预后不良相关,其中 ACTG1 表达与预后关联稳定一致,被确定为核心剪接调控因子。
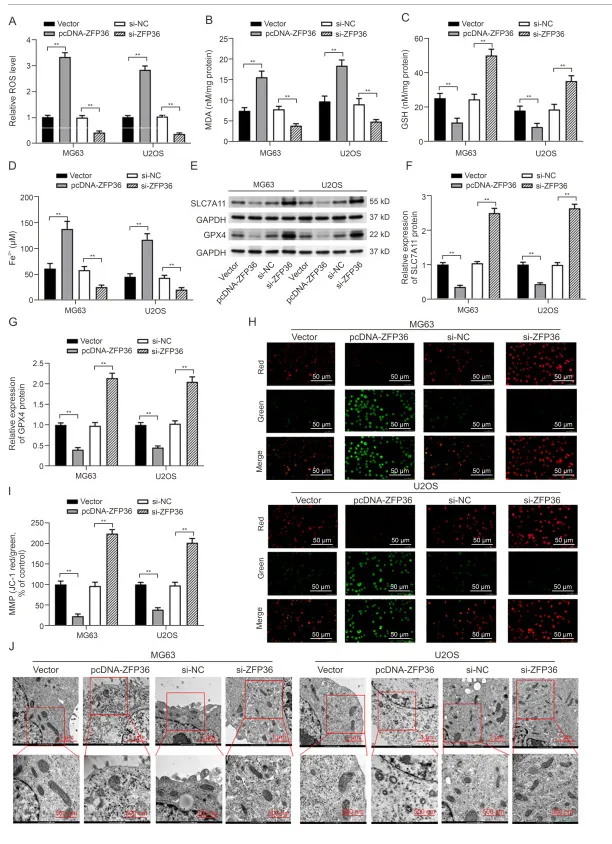
ZFP36 过表达加剧了 OS 细胞中的铁消亡和线粒体功能障碍
ZFP36 为铁死亡相关基因。过表达 ZFP36 可升高 ROS、MDA 及 Fe²⁺水平,降低 GSH 含量,抑制 GPX4、SLC7A11 表达,并破坏线粒体膜电位、造成线粒体结构损伤;敲低 ZFP36 则作用相反。提示 ZFP36 过表达可促进骨肉瘤细胞铁死亡与线粒体功能障碍。
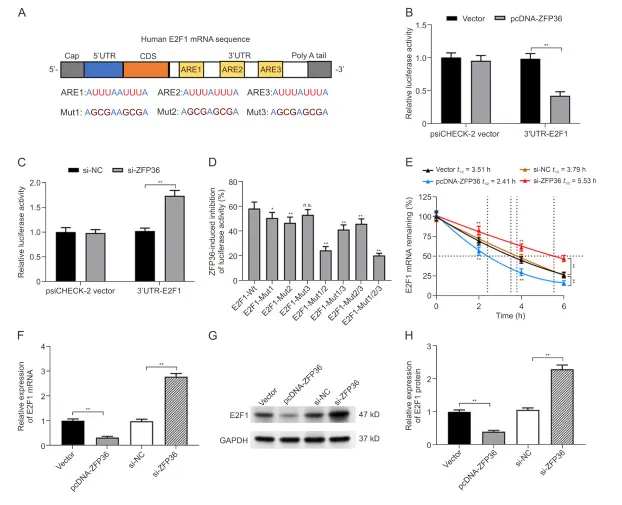
ZFP36通过促进O2F1 mRNA在OS细胞中的降解来抑制E2F1表达
ZFP36 可结合转录本 3ʹUTR 的 AREs 促进 mRNA 降解。E2F1 mRNA 3ʹUTR 含 3 条 ARE 序列,ZFP36 过表达可降低其荧光素酶活性,敲低则相反;突变 ARE1 和 ARE2 可逆转该抑制效应,ARE3 无影响。放线霉素 D 实验显示,ZFP36 过表达缩短 E2F1 mRNA 半衰期、加速其降解,同时抑制 E2F1 mRNA 及蛋白表达,敲低则作用相反。
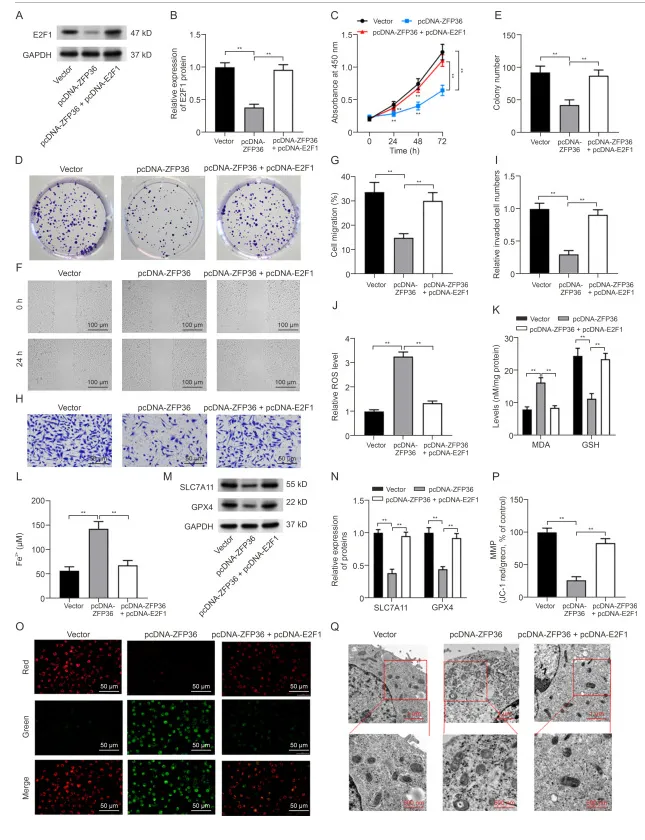
ZFP36过表达通过抑制E2F1表达,促进铁减脂和线粒体功能障碍,抑制了OS细胞的进大
在 MG63 细胞中,ZFP36 过表达可降低 E2F1 蛋白水平,pcDNA-E2F1 转染则可升高 E2F1 表达。功能挽救实验显示,E2F1 过表达可逆转 ZFP36 过表达介导的细胞增殖、克隆形成、迁移和侵袭抑制,以及凋亡促进作用;同时可逆转 ZFP36 过表达引起的 ROS、MDA、Fe²⁺水平升高,GSH 水平下降,GPX4、SLC7A11 表达抑制,以及线粒体膜电位损伤和结构异常。
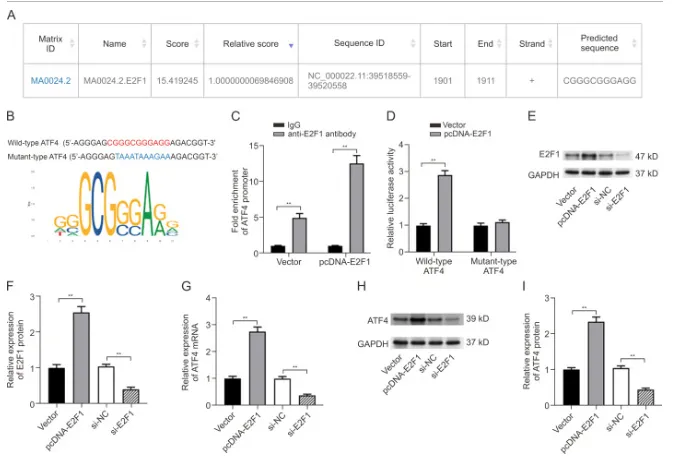
E2F1促进ATF4的转录活化
已知 ATF4 在多种癌症中可调控铁死亡,通过 Jaspar 数据库预测发现 ATF4 启动子存在 E2F1 结合位点;ChIP 实验证实 E2F1 可富集于 ATF4 启动子,且 E2F1 过表达仅能提升野生型 ATF4 的荧光素酶活性,对突变型无影响。此外,E2F1 过表达可提高 MG63 细胞中 ATF4 的 mRNA 和蛋白水平,敲低 E2F1 则抑制 ATF4 表达,表明 E2F1 可通过转录激活促进 ATF4 表达。
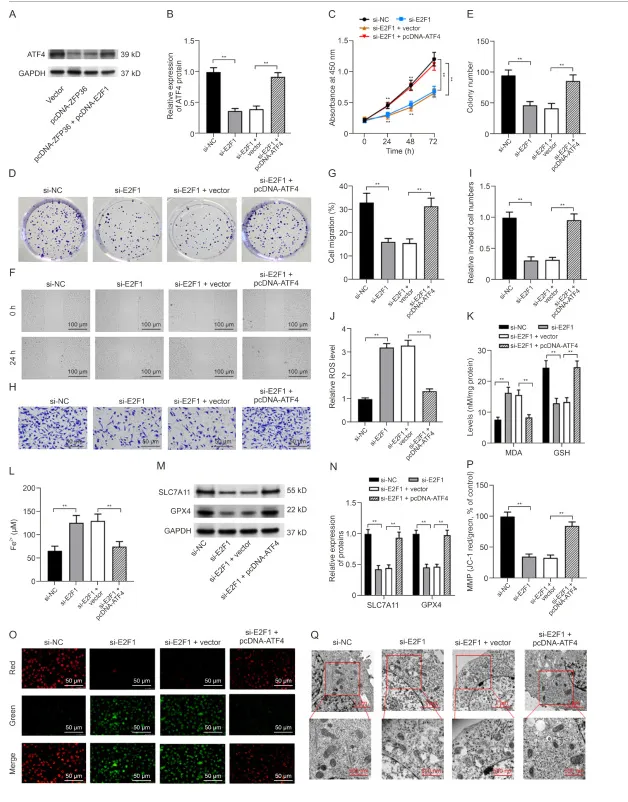
E2F1敲低通过降低ATF4表达促进铁磷作用和线粒体功能障碍,抑制OS细胞的进展
在 MG63 细胞中,敲低 E2F1 可降低 ATF4 表达,而过表达 ATF4 则可逆转这一效应。功能回复实验显示,ATF4 过表达能够抵消 E2F1 敲低所导致的细胞增殖、克隆形成、迁移及侵袭抑制,并可逆转其诱导的 ROS、MDA、Fe²⁺升高、GSH 下降、GPX4/SLC7A11 变化,以及线粒体膜电位损伤与结构异常。
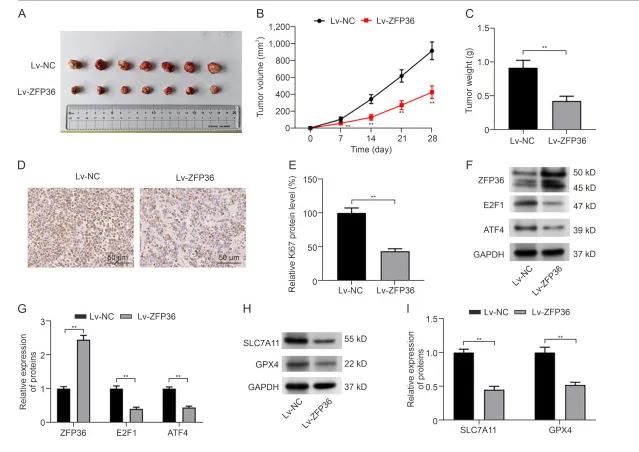
ZFP36过表达抑制了小鼠的OS肿瘤生长
构建骨肉瘤异种移植瘤模型后发现,ZFP36 过表达组肿瘤体积与重量均显著降低。IHC 结果显示,该组 Ki67、E2F1、ATF4、GPX4 及 SLC7A11 蛋白水平均下降,证实 ZFP36 过表达在体内可显著抑制骨肉瘤生长。
【文章小结】
本研究证实 ZFP36 在 OS 中发挥抑癌作用,通过靶向降解 E2F1、抑制 ATF4 转录,诱导 OS 细胞铁死亡及线粒体功能障碍,抑制恶性进展。研究阐明 ZFP36/E2F1/ATF4 轴调控机制,为 OS 靶向治疗提供理论依据与新靶点。
如果你也想用同款思路分析,需要生信分析,复现高分思路的同学,快关注后台咨询吧!生信解码站,助力每一个科研梦想!









 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
