通讯作者
朱云峰(南京工业大学)
研究背景
氢能作为清洁能源载体,在能源转型中具有重要地位。固态储氢技术,特别是镁基储氢合金,因其低成本、高储氢容量和良好的可逆性而备受关注。然而,氢化镁(MgH2)的商业化应用受限于其缓慢的动力学性能和较高的操作温度。近年来,催化剂添加和合金化策略被证明是改善MgH2储氢性能的有效方法。其中,TiO2因其优异的催化活性、低毒性和高稳定性而受到青睐,而Ni在提升MgH2脱氢动力学方面表现出卓越的催化效果。本研究通过设计双相TiO2负载Ni纳米颗粒催化剂,结合Mg90Al10合金体系,探索协同催化策略对MgH2储氢性能的增强机制。
痛点问题
1. MgH2动力学性能缓慢,导致吸放氢速率低,需要较长时间完成储氢过程
2. MgH2操作温度高,起始脱氢温度通常超过300°C,限制了实际应用场景
3. 单一催化剂难以同时优化动力学和热力学性能,需要开发协同催化策略
4. 催化剂在循环过程中易发生团聚或失活,影响长期稳定性
5. MgH2的热力学稳定性高,脱氢焓值大,导致储氢效率降低
6. 缺乏对催化剂-基体界面电子相互作用的深入理解,限制了催化剂设计优化
核心发现
1. 成功制备了双相TiO2(锐钛矿/金红石)负载Ni纳米颗粒催化剂(Ni/Dp-TiO2),Ni颗粒尺寸为5-15 nm,均匀分散在双相TiO2载体上
2. Mg90Al10-10 wt.% Ni/Dp-TiO2复合材料展现出优异的储氢性能:150°C下2500 s内吸收4.28 wt.% H2,250°C下2500 s内脱附3.86 wt.% H2,显著优于Mg90Al10基体材料
3. 起始脱氢温度从Mg90Al10的325°C降低至182°C,降低了143°C,展现出显著的动力学优势
4. 脱氢活化能从Mg90Al10的155.95 kJ/mol大幅降低至56.36 kJ/mol,表明催化效果显著
5. 氢化焓和脱氢焓分别降低至-66.25和68.18 kJ/mol,相比纯MgH2的-76.24和80.86 kJ/mol有明显改善
6. 通过XRD和XPS分析证实,在球磨和循环过程中原位形成了AlNi金属间化合物,与Dp-TiO2和Al物种(Mg-Al固溶体、Mg17Al12、Mg2Al3)协同作用
7. DFT计算揭示了AlNi/Dp-TiO2界面的电子调控效应,优化了功函数和氢吸附能,是性能提升的关键机制
8. 材料在20次循环后容量保持率达99.8%,展现出优异的循环稳定性
图文解析
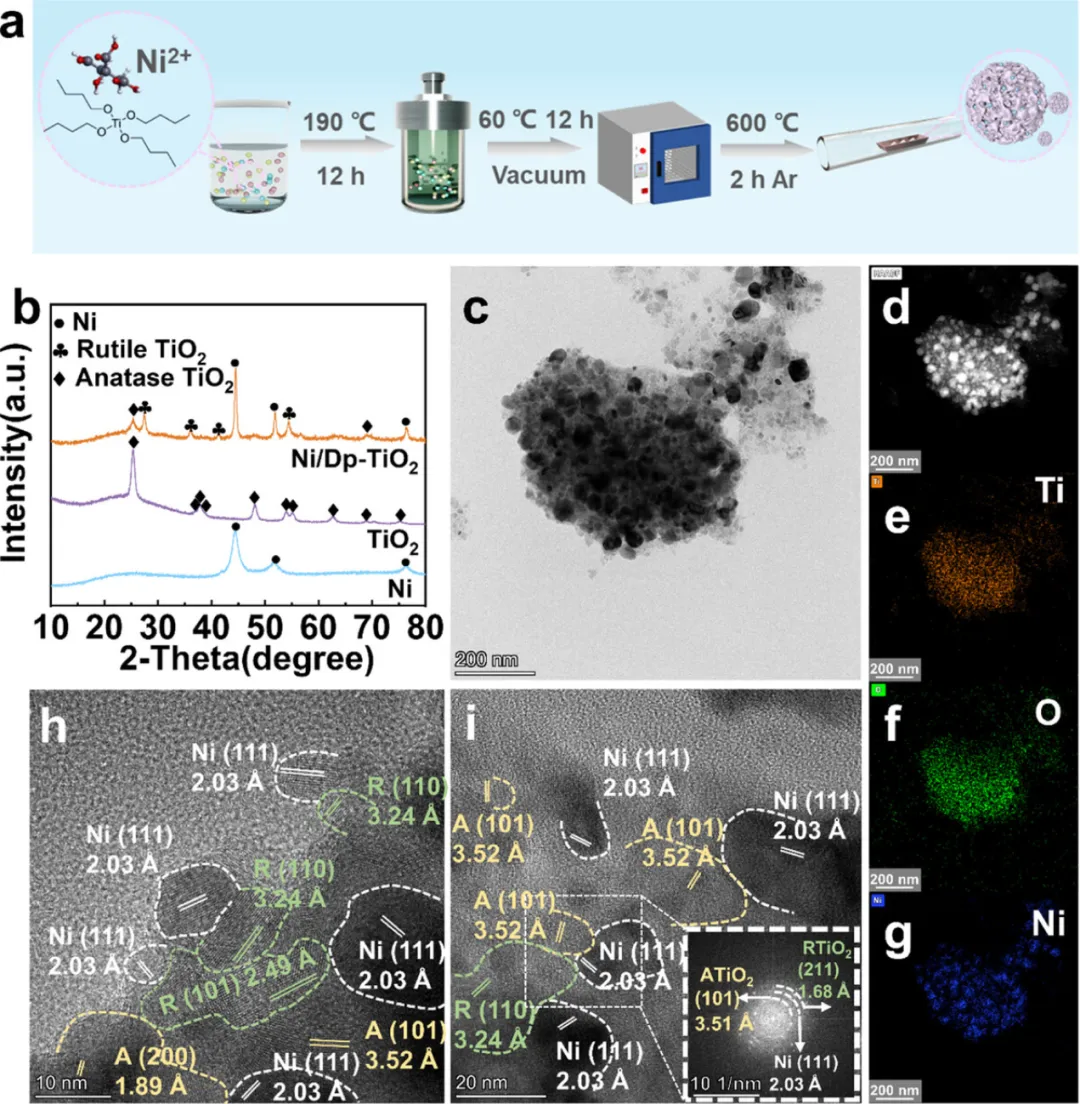
图1:Ni/Dp-TiO2催化剂的制备与形貌表征
1. 图1a展示了Ni/Dp-TiO2的水热法制备流程示意图,包括TiO2载体制备和Ni负载两个关键步骤
2. XRD图谱(图1b)显示Ni以元素态存在,TiO2在Ni负载后转变为锐钛矿和金红石双相结构,质量比接近1:1
3. TEM和HAADF-STEM图像(图1c-d)揭示了Ni纳米颗粒(5-15 nm)在Dp-TiO2载体上的均匀分散
4. EDS元素分布图(图1e-g)证实了Ni、Ti、O元素的均匀分布,表明催化剂的成功制备
5. HRTEM图像(图1h-i)清晰显示了Ni(111)、锐钛矿TiO2(101)和金红石TiO2(110)的晶面间距,分别为2.03、3.52和3.24 Å
6. FFT衍射花样证实了Ni颗粒与Dp-TiO2之间存在紧密的相界面,有利于电子传输
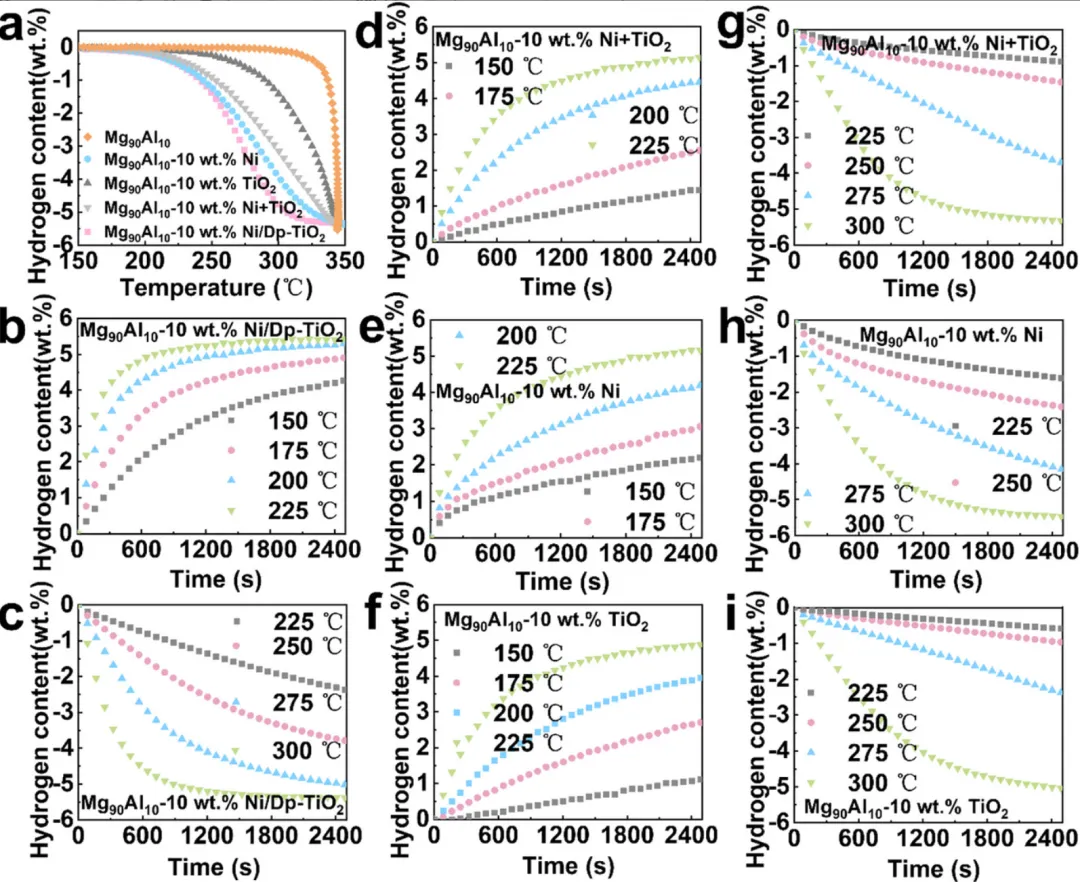
图2:Mg90Al10复合材料的储氢性能测试
1. TPD曲线(图2a)显示Mg90Al10-10 wt.% Ni/Dp-TiO2的起始脱氢温度为182°C,比Mg90Al10降低了143°C
2. Ni/Dp-TiO2催化剂的性能优于Ni+TiO2机械混合物,表明存在协同催化效应
3. 等温吸氢曲线(图2b, d-f)显示Mg90Al10-10 wt.% Ni/Dp-TiO2在150°C下2500 s内吸收4.28 wt.% H2
4. 等温脱氢曲线(图2c, g-i)显示在250°C下2500 s内脱附3.86 wt.% H2
5. 对比样品(Mg90Al10-10 wt.% Ni、Mg90Al10-10 wt.% TiO2、Mg90Al10-10 wt.% Ni+TiO2)的性能均不如Ni/Dp-TiO2复合材料
6. Mg90Al10基体材料在相同条件下仅能吸收0.96 wt.% H2和脱附0.27 wt.% H2,性能差距显著
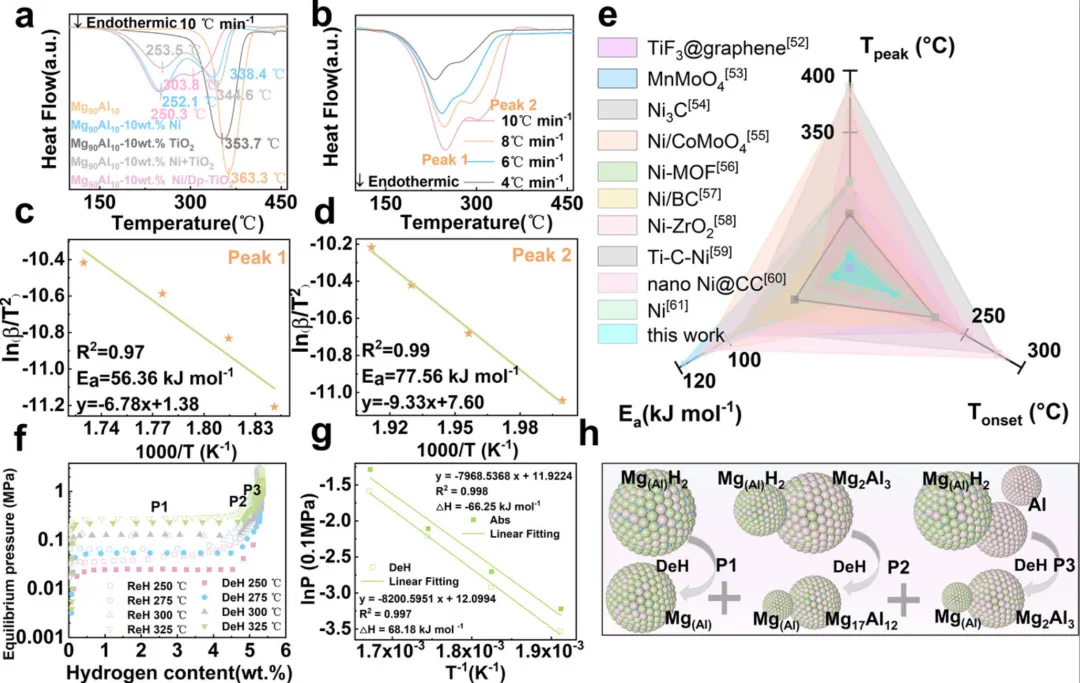
图3:脱氢动力学与热力学性能分析
1. DSC曲线(图3a)显示Mg90Al10-10 wt.% Ni/Dp-TiO2的脱氢峰温为250.3°C和303.8°C,均低于Mg90Al10的363°C
2. 通过Kissinger方程计算得到脱氢活化能为56.36和77.56 kJ/mol(图3b-d),显著低于Mg90Al10的155.95 kJ/mol
3. 图3e对比了多种MgH2-催化剂体系的起始脱氢温度、峰温和活化能,Ni/Dp-TiO2展现出最优性能
4. PCT曲线(图3f)在250-325°C范围内显示了三个平台,对应不同的相变过程
5. Van't Hoff图(图3g)确定了氢化焓(-66.25 kJ/mol)和脱氢焓(68.18 kJ/mol),相比纯MgH2有明显降低
6. 图3h示意图展示了脱氢过程中的相变反应:P1、P2、P3平台分别对应Mg(Al)H2分解为Mg(Al)+H2、Mg(Al)+Mg17Al12+H2、Mg(Al)+Mg2Al3+H2
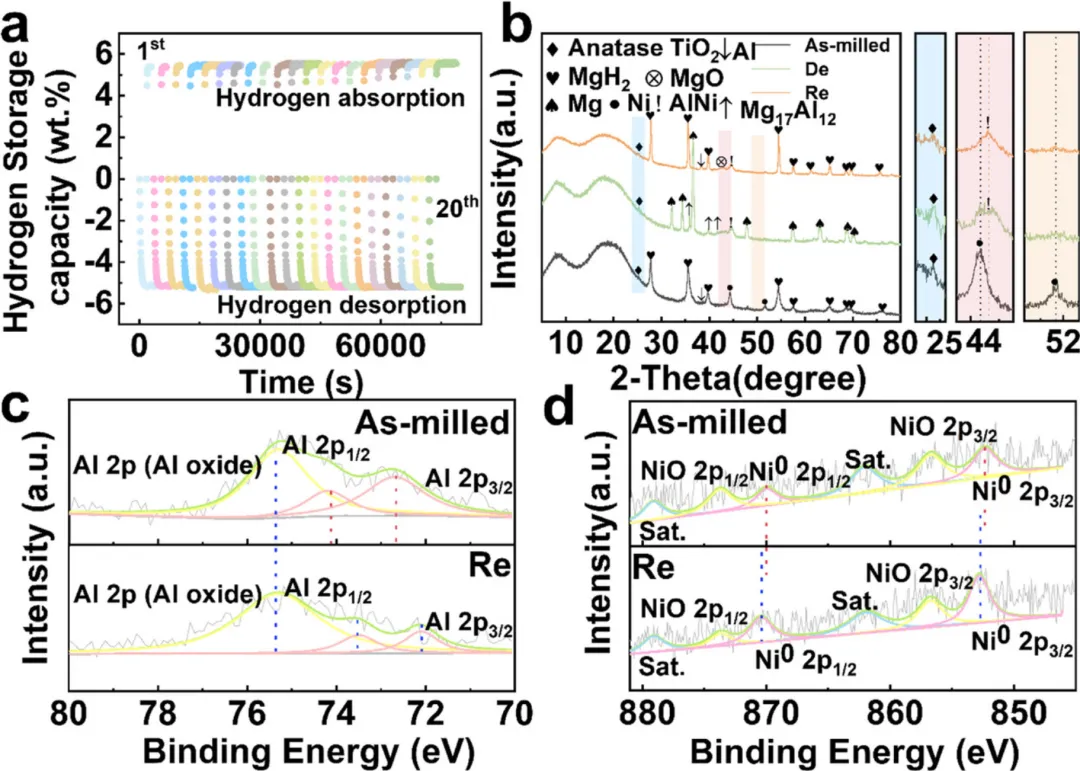
图4:循环稳定性与相结构演变
1. 20次循环测试(图4a)显示Mg90Al10-10 wt.% Ni/Dp-TiO2的容量保持率达99.8%,表明优异的循环稳定性
2. XRD图谱(图4b)分析了球磨态、脱氢态和再氢化态的相结构演变
3. 球磨态样品主要包含MgH2、Al、TiO2和Ni相
4. 脱氢后出现Mg、Mg17Al12、AlNi等新相,表明发生了相转变
5. 再氢化后MgH2相恢复,AlNi相保持稳定,证实了催化剂的结构稳定性
6. XPS分析(图4c-d)显示Al 2p和Ni 2s的结合能变化,揭示了界面电子相互作用
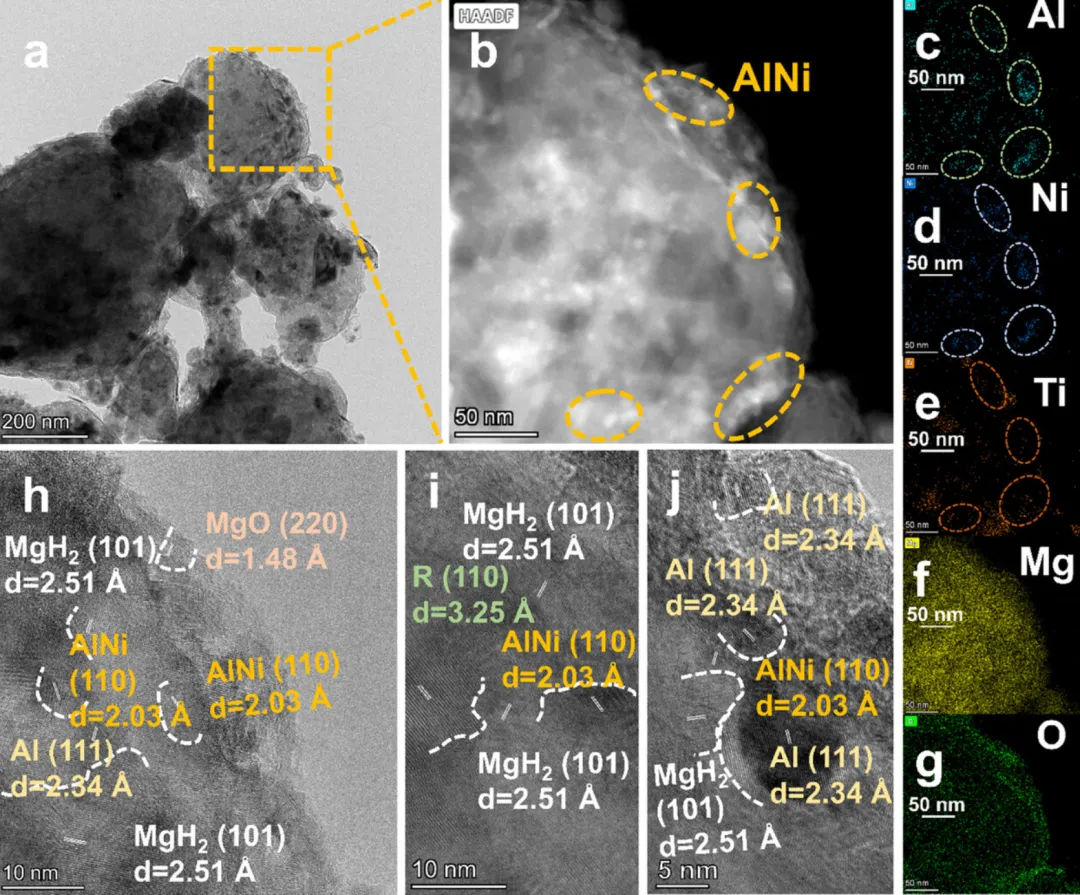
图5:再氢化后材料的微观结构表征
1. TEM和HAADF图像(图5a-b)显示再氢化后材料仍保持良好的纳米结构
2. EDS元素分布图(图5c-g)证实Ni、Ti、O、Mg、Al元素的均匀分布
3. HRTEM图像(图5h-j)清晰显示了MgH2、AlNi、TiO2等相的晶格条纹
4. AlNi相的存在证实了其在循环过程中的稳定性,是催化活性的关键
5. 界面结构保持完整,有利于持续的电子调控效应
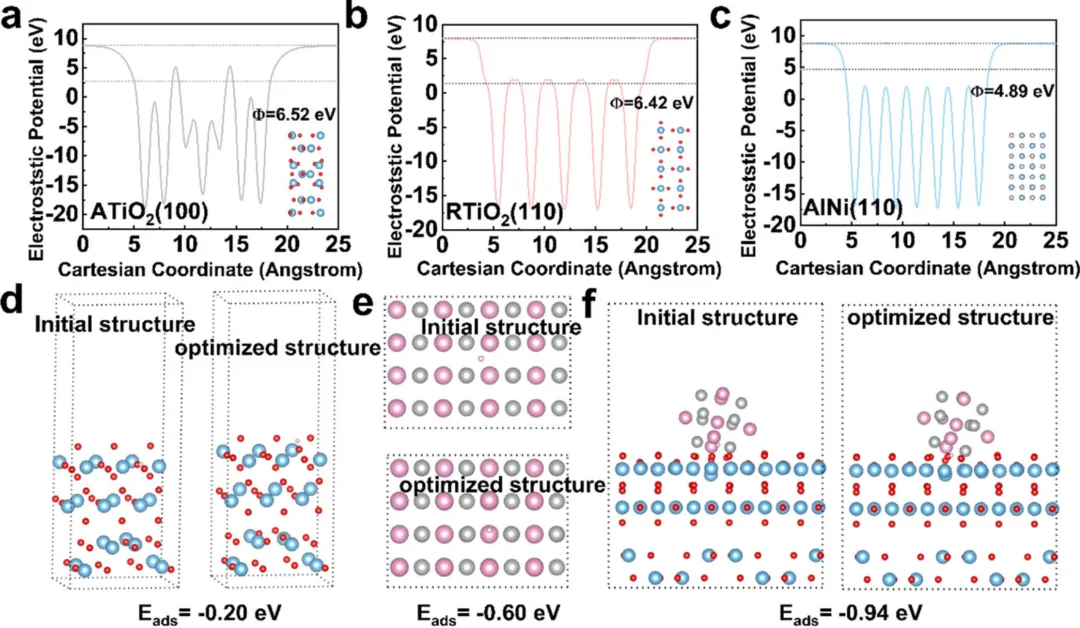
图6:DFT计算揭示催化机理
1. 静电势计算(图6a-c)显示了锐钛矿TiO2(100)、金红石TiO2(110)和AlNi(110)表面的电子分布特征
2. AlNi/Dp-TiO2界面的功函数优化,促进了电子转移和氢吸附
3. 氢吸附能计算揭示了AlNi/Dp-TiO2界面是氢吸附的活性位点
4. 电子密度差分析证实了AlNi与TiO2之间的强电子相互作用
5. 理论计算结果与实验观察一致,阐明了协同催化的电子机制
原文链接
https://doi.org/10.1002/smll.73509