化学反应网络的自动化搜索对于反应机理解析及新反应(或催化剂)设计至关重要。然而,复杂反应过程往往涉及大量中间体、过渡态及多条竞争反应路径,传统量子化学方法计算量大,难以支撑自动化的高效探索。反应性机器学习势(MLP)在精度与效率之间能取得平衡,但目前已报道的高精度、通用反应性MLP仍然有限,其发展主要受制于高质量反应性数据集的高效构建。
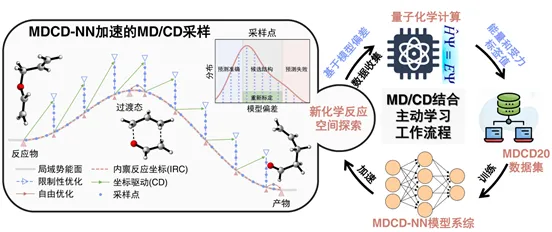
近日,南京大学化学学院黎书华、王国强与绿色化学与工程研究院杨满意研究团队,基于前期发展的分子动力学结合坐标牵引(MD/CD)反应路径自动搜索方法,提出了MD/CD-主动学习(MD/CD-AL)框架(图1),实现高质量反应性数据集的高效、自动化采集。基于该框架,团队构建了H/C/N/O元素的百万级规模的数据集MDCD20,并训练得到反应性机器学习势MDCD-NN。该势函数在计算精度上与参考的密度泛函理论(DFT)方法相当,使MD/CD的计算效率提升了四个数量级及以上,为复杂体系的反应机理自动化探索开辟了新路径。相关研究成果已发表于《中国化学会化学》CCS Chemistry期刊。
图1. MD/CD-AL框架示意图。
(一)数据集构建与模型表现
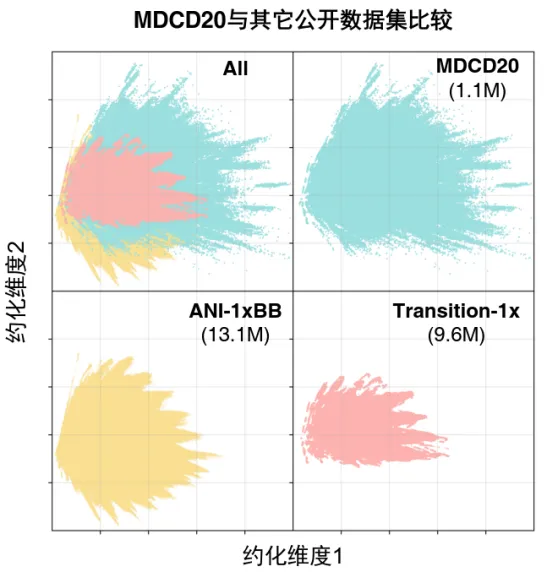
为解决高质量反应性数据稀缺的核心瓶颈,南京大学团队将MD/CD与主动学习策略结合,以MD/CD方法主动采样反应空间中未充分覆盖的过渡态、中间体等非平衡结构,再训练MDCD-NN加速MD/CD计算,最终实现了关键反应性结构的高效获取。经过50轮主动学习迭代,团队构建了包含约140万个非平衡结构的数据集MDCD20,包括中性闭壳层分子、自由基分子等,涉及H/C/N/O元素。尽管MDCD20的数据量仅为部分同类公开数据集(如Transition-1x、ANI-1xBB等H/C/N/O数据集)的十分之一左右,但其化学空间覆盖范围更为广泛(图2)。
图2. MDCD20与ANI-1xBB和Transition-1x数据集的化学空间覆盖范围比较(M表示百万)。图中各结构在化学空间中的坐标由其 SOAP(Smooth Overlap of Atomic Positions)描述符表示,并通过 PCA(Principal Component Analysis)降维至二维。
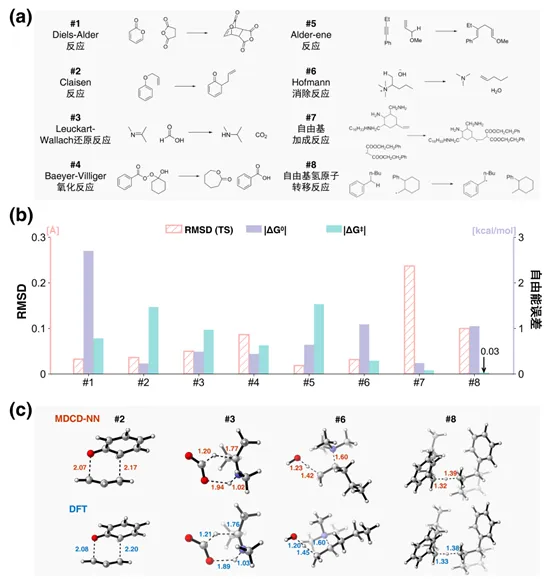
基于该数据集训练的MDCD-NN机器学习势,在181个教科书级基元反应的过渡态定位任务中实现了100%的成功率。其预测的过渡态结构与参考DFT(gCP(TZ)-ωB97X-D4/def2-TZVP)之间的RMSD偏差不超过0.2 Å;对反应Gibbs自由能变与势垒预测的平均绝对误差(MAE)均低于2 kcal/mol。部分反应的测试结果如图3所示。
图3. MDCD-NN与其参考DFT在8个基元反应上的自由能和结构比较。(a) 所考察的基元反应; (b) MDCD-NN相比于DFT的误差,包括过渡态结构的RMSD值,反应Gibbs自由能变(∆G0)和自由能垒(∆G‡)的绝对误差;(c) 示例反应过渡态结构对比。
(二)典型应用示例:复杂反应网络与动态自由能面
为验证MDCD-NN在真实复杂体系反应网络中的精度与效率,研究团队进一步利用MD/CD自动搜索框架开展了系统评估。验证的体系包括:文多灵级联反应、脯氨酸催化的不对称Aldol反应以及AIBN引发的自由基级联反应等。结果表明,该模型具备优异的可迁移性,其预测精度可与参考DFT计算相当,计算效率则提升了四个数量级以上。
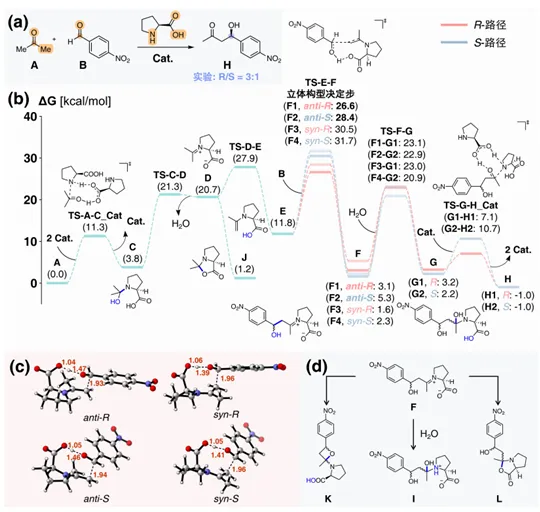
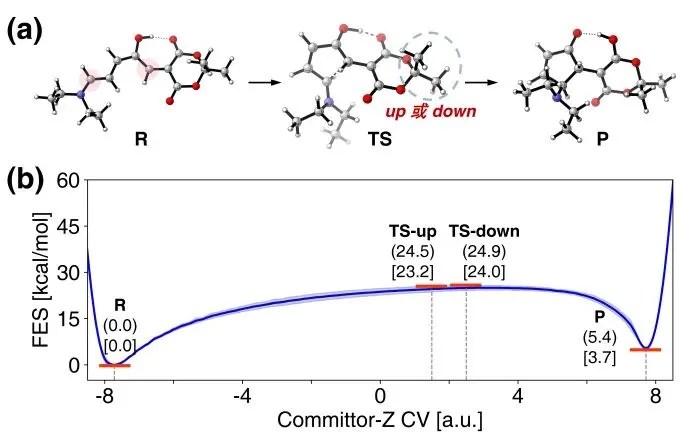
以List等人发展的脯氨酸催化的不对称Aldol反应为例(图4):在微观层面上,该反应过程涉及多分子参与、构象高度柔性的催化循环。要精确预测反应的立体选择性,要求方法兼具反应网络的完备性与计算精度。MDCD-NN加速的MD/CD仅需调用一张V100 GPU计算80小时,即可得到包含近800个中间体构象和600个过渡态构象的总反应网络,其中最优催化循环由6步基元反应组成。在该循环中,MDCD-NN成功定位了决定对映选择性的关键过渡态,预测的立体选择性∆∆G‡与DFT结果高度吻合(1.8 vs 1.9 kcal/mol),并自动搜索到了传统方法难以计算过程中被忽略的“双分子协同催化”路径(TS-A-C_Cat、TS-G-H_Cat)。
图4. MDCD-NN加速的MD/CD在脯氨酸催化的不对称Aldol反应中的应用。(a) 反应通式。(b) 最优催化循环的自由能曲线图。(c) 立体构型决定步TS-E-F的四种结构。(d) 其它循环外反应路径途径示例。
图5. MDCD-NN在纳秒级增强采样动力学模拟的应用。(a) 总包反应式。(b) Reweighting后的自由能面,灰色阴影表示标准差。
MDCD-NN不仅可用于能量极小结构的优化与分析,还能适用于动态反应过程的模拟。虽然训练集MDCD20的结构主要来源于能量极小化轨迹,模型仍可稳定支持纳秒级增强采样分子动力学模拟,准确重现供体-受体Stenhouse加合物(DASA)的电环化反应自由能面(图5),进一步验证了其在复杂动力学场景中普适性。
总结与展望
传统DFT方法在处理类似化学反应过程时,单次反应网络构建往往需要数月甚至更长时间;而MDCD-NN将这一过程缩短至数天,同时保持DFT水平的计算精度。这一方法使针对复杂有机反应、酶催化及材料降解等复杂反应过程的系统性机理探索成为可能。目前,MDCD-NN的适用范围仍限于H、C、N、O四种元素。研究团队计划在后续研究中,将MD/CD-AL工作流扩展至B、P、S、F等更多元素体系,并引入长程静电校正与解析Hessian等新功能,以进一步提升模型的普适性与能力,推动从“算得动”向“算得更准、更快”的演进。
相关研究成果发表在CCS Chemistry杂志上,该论文的第一作者为南京大学博士生李国奥,通讯作者为南京大学化学学院黎书华教授、王国强副教授和绿色化学与工程研究院杨满意副教授。课题组凌皓博、苏朝旭、刘正轩同学参与了本工作的研究。该研究得到了国家重点研发计划、国家自然科学基金、新基石科学基金会及江苏省前沿项目的支持。
原文(扫描或长按二维码,识别后直达原文页面):

A data-efficient reactive machine learning potential to accelerate automated exploration of complex reaction networks
Guoao Li, Haobo Ling, Chaoxu Su, Zhengxuan Liu, Guoqiang Wang*, Manyi Yang*, Shuhua Li*
CCS Chem., 2026, DOI: 10.31635/ccschem.026.202607339
导师介绍
黎书华
https://itcc.nju.edu.cn/~shuhua/index_cn.html
https://www.x-mol.com/university/faculty/410629
王国强
https://www.x-mol.com/groups/wang_guoqiang

杨满意
https://frontier.nju.edu.cn/86/07/c59289a689671/pagem.htm
https://www.x-mol.com/university/faculty/415238
点击“阅读原文”,查看 化学 • 材料 领域所有收录期刊



 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
