南京工业大学AFM:路易斯酸和PO43-修饰赋予锚定于磷掺杂RuO2纤维气凝胶上的Ni/Cr2O3在碱性海水电解中的双重抗腐蚀机制
1、文章亮点
1. 该研究通过静电纺丝结合金属颗粒自组装技术,成功构筑了由Ni/Cr2O3纳米颗粒锚定在P-RuO2纳米纤维上的三维分级多孔气凝胶。该结构的高比表面积(54.2 m2g-1)和丰富的介孔/大孔网络不仅暴露了大量催化活性位点,还促进了反应物扩散与电子传输,从而在碱性海水中实现了优异的OER/HER双功能催化活性,过电位低至220/27 mV@10 mA cm-2)。
2. 揭示了Cr2O3作为路易斯酸选择性富集OH-形成局部碱性微环境,同时P掺杂RuO2在OER过程中原位生成PO43-并通过静电排斥作用阻挡Cl-腐蚀。这种协同机制有效抑制了RuO2的过氧化失活和氯离子侵蚀,使AEMSE器件在0.1 A cm-2下稳定运行500小时,且电压衰减仅60 µV h-1,突破了海水电解催化剂的耐久性瓶颈。
2、全文速览
阴离子交换膜海水电解(AEMSE)对于未来大规模绿氢生产至关重要,但目前面临缺乏高耐久性析氧反应(OER)电催化剂的挑战。该研究报道了一种由锚定在磷掺杂二氧化钌纳米纤维上的镍/氧化铬纳米颗粒(Ni/Cr2O3@P-RuO2)组成的多孔气凝胶。该Cr2O3@P-RuO2气凝胶在碱性海水中对OER和析氢反应(HER)分别表现出220 mV和27 mV@10 mA cm-2的低过电位,并且在组装到AEMSE中后,能够在0.1 A cm-2下稳定运行500小时。X射线吸收近边结构(XANES)分析结合密度泛函理论(DFT)计算共同揭示,多金属协同效应诱导电子从Ni/Cr2O3向Ru转移。此外,Ru表现出作为催化活性位点的不饱和配位缺陷,从而增强了催化活性。原位拉曼光谱和飞行时间二次离子质谱(TOF-SIMS)证实催化剂表面形成了PO43-,通过静电排斥作用提高了耐腐蚀性。另外,高度分散的Cr2O3颗粒阻止了RuO2在OER过程中的过氧化和失活。同时,它们作为路易斯酸位点,与PO43-协同作用,形成对OH-具有高选择性的表面微环境。分子动力学模拟验证了这种双重抗腐蚀机制的建立,为解决海水电解过程中催化剂耐久性瓶颈问题实现了突破。
3、背景介绍
开发以可再生能源为驱动的绿色氢能载体,对于实现可持续未来至关重要。虽然基于可再生能源的电解水制氢技术高效、环保且操作简便,但该过程消耗大量纯水,难以用于大规模制氢。而海水能够缓解日益严峻的淡水资源短缺问题。电催化水分解作为一种绿色高效的方法备受关注,其中包括阴极HER和阳极OER。其中,阴离子交换膜水电解(AEMWE)技术能够在弱碱性体系中运行,兼具碱性水电解的成本优势和对间歇性可再生电力的适应性,因此在绿氢生产中极具应用前景。基于此,发展阴离子交换膜海水电解(AEMSE)技术具有重要的现实意义和紧迫性。然而,目前AEMSE的研究进展仍然有限,主要原因是海水中高浓度的氯离子(Cl-)不仅会引发竞争的析氯反应(ClER),降低OER选择性,还会造成催化剂表面腐蚀,破坏催化剂结构,导致性能急剧下降。因此,开发兼具高活性、高选择性和强耐Cl-腐蚀能力的析氧阳极电催化剂,已成为实现高效海水电解的关键技术。
铂族催化剂因其优异的催化活性而被广泛使用,但其高昂成本和稀缺性限制了大规模应用。在众多有前景的候选材料中,钌基催化剂因其出色的电催化性能、相对较低的成本和良好的化学稳定性而备受关注。Ru独特的d轨道电子结构赋予其本征OER催化活性,其适中的氢吸脱附行为有效降低了HER的能垒。然而,海水中高浓度的Cl-会与Ru反应生成可溶性盐,使RuO2转化为不稳定且可溶的高价态物种(Run+,n > 4),导致Ru基催化剂在海水中更易腐蚀失活。根据近期研究,设计耐腐蚀催化剂的策略可归纳为六种:(1)多聚阴离子静电排斥层;(2)富路易斯酸的OH-微环境;(3)致密的物理阻挡层;(4)动态自修复层;(5)晶格保护设计。其中,表面保护层的形成已成为抗氯化物腐蚀的主要策略,且这一研究方向正从单一物理屏障向多机制协同作用拓展。此外,根据Pearson的硬软酸碱(HSAB)原理,硬酸优先与硬碱结合。因此,增强电催化剂的路易斯酸性可以有效促进OH-富集并提高OER选择性。将这些抗腐蚀策略协同应用于本征活性的Ru基催化剂上,为开发高效海水电解催化剂提供了一条潜在途径。然而,迄今为止报道的大多数Ru基电催化剂呈现一维或二维结构。为了进一步提高比表面积以暴露更多活性位点,并防止RuO2发生过氧化和腐蚀失活,开发具有三维多孔结构的RuO2基催化剂具有重要意义。
气凝胶的三维多孔自支撑结构作为水电解催化剂表现出显著优势。这归因于气凝胶连续的三维骨架形成了高效导电通路,缩短了电子传输距离并显著降低了电阻损耗。此外,气凝胶的分级多孔结构极大增加了活性位点的暴露,远超传统团聚颗粒催化剂。然而,气凝胶在AEMSE中的应用研究仍然相对较少。将气凝胶与其他活性物种耦合构建异质结构是一种有前景的策略,但实现用于增强耐腐蚀性和OER性能的电子结构调控仍然是一个挑战。
该研究报道了一种通过静电纺丝结合金属颗粒自组装合成的三维多孔气凝胶,该气凝胶由锚定在P-RuO2纳米纤维上的Ni/Cr2O3纳米颗粒组成(Ni/Cr2O3@P-RuO2)。所得催化剂在海水电解中对碱性HER和OER均表现出双功能催化性能。Ni/Cr2O3@P-RuO2气凝胶由直径约100 nm的P-RuO2纤维组成,其表面均匀锚定Ni和Cr2O3纳米颗粒。该结构含有大量介孔和大孔,分级孔隙率有利于中间体的高效扩散和活性位点的充分暴露。最优的Ni/Cr2O3@P-RuO2气凝胶在碱性海水中表现出优异的OER/HER性能,在10 mA cm-2下的过电位分别低至220 mV和27 mV。当应用于AEMSE器件时,所制备的催化剂在2.0 V电压下达到1.5 A cm-2的高电流密度,并在0.1 A cm-2下连续运行500小时无明显性能衰减。XANES分析结合DFT计算共同揭示,多金属协同效应诱导了电子从Ni/Cr2O3向Ru转移。此外,Ru作为催化活性位点表现出不饱和配位缺陷,从而增强了OER/HER活性。原位电化学拉曼光谱揭示了催化剂表面形成了羟基氧化物,同时生成了能够通过静电相互作用排斥Cl-的PO43-。晶体轨道重叠布居(COOP)分析进一步表明,Ru与*OOH之间的强键合效应增强了对中间体的吸附能力,从而加速了OER动力学。此外,高度分散的纳米级Cr2O3颗粒有效防止了RuO2在OER过程中的过氧化和失活。这种双重抗腐蚀机制的协同效应有效增强了催化剂的耐氯性,突破了在高盐海水环境中催化剂稳定性差的瓶颈,实现了在工业电流密度下的海水电解。4、图文解析
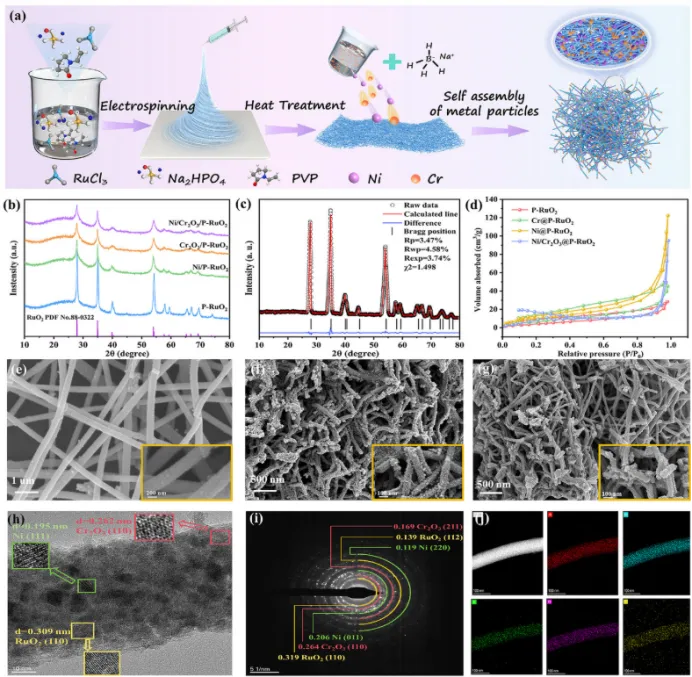
图1. (a) Ni/Cr2O3@P-RuO2气凝胶的制备流程示意图;(b) 样品的XRD图;(c) Ni/Cr2O3@P-RuO2气凝胶XRD的Rietveld精修结果;(d) 所得样品的氮气吸附-脱附等温线;(e) P-RuO2纤维、(f) Ni@P-RuO2气凝胶、(g) Ni/Cr2O3@P-RuO2气凝胶的SEM图;(h) HRTEM图、(i) SAED图、(j) Ni/Cr2O3@P-RuO2的EDS元素分布图。
采用静电纺丝结合煅烧的方法合成了P-RuO2纳米纤维膜,其中以RuCl3·3H2O、Na2HPO4和聚乙烯吡咯烷酮分别作为Ru源、P源和成纤剂。随后,通过金属颗粒自组装技术将Ni2+和Cr3+负载到P-RuO2纳米纤维表面。这一过程通过颗粒间结合力促进了P-RuO2纳米纤维内部三维多孔网络的形成。最终,通过冰晶导向生长和冷冻干燥技术,将这些纤维组装成具有分级多孔结构的Ni/Cr₂O₃@P-RuO2气凝胶(图1a)。图1b展示了Ni/Cr2O3@P-RuO2气凝胶及对照样品的XRD图。值得注意的是,负载Ni和Cr2O3后气凝胶样品的衍射峰强度均有所减弱。然而,未观察到归属于Ni和Cr物种的衍射峰,表明它们可能以高度分散的纳米尺度形态存在且结晶度较低。此外,我们对Ni/Cr2O3@P-RuO2的XRD谱图进行了Rietveld精修,结果显示所有衍射峰均对应于金红石相RuO2(空间群:P42/mnm)(图1c)。为了研究气凝胶样品的多孔结构,通过氮气吸附/脱附等温线分析了所得样品的孔结构(图1d)。所有样品均表现出混合的II型和IV型吸附等温线,在相对压力0.99处未观察到饱和平台,表明样品中存在丰富的大孔。此外,图中由毛细凝聚产生的滞后环证实了样品中介孔的存在。因此,气凝胶样品呈现出由介孔和大孔组成的分级孔结构。值得注意的是,这种三维分级多孔网络有助于Ni/Cr2O3@P-RuO2气凝胶增大比表面积,促进了活性位点的暴露,增强了催化剂与反应物的接触和扩散,从而提升了OER活性。SEM图表明,所制备的气凝胶样品由直径约200 nm的卷曲纤维组成(图1e)。对于Ni@P-RuO2样品,可以观察到Ni颗粒交联形成网络结构,均匀锚定在纤维表面(图1f)。最优的Ni/Cr2O3@P-RuO2气凝胶样品呈现出由直径约30–50 nm的介孔和超过100 nm的大孔组成的分级孔结构(图1g)。大孔有利于析氧过程中的传质,而介孔使气凝胶网络能够暴露大量催化活性位点。因此,构建这种分级多孔结构提高了催化活性。进一步采用TEM分析了最优Ni/Cr2O3@P-RuO2气凝胶的晶体微观结构和形貌。图1h显示了由直径2–10 nm的Ni/Cr2O3颗粒锚定的纤维结构。此外,对HRTEM图像进行FFT处理,显示出0.309 nm的晶格间距,对应于RuO2的(110)晶面;而0.309 nm和0.262 nm的晶格间距分别归属于Ni的(111)晶面和Cr2O3的(110)晶面。另外,SAED图像呈现出清晰的多晶衍射环特征,分别对应于RuO2、Ni和Cr2O3的晶面(图1i)。Ni/Cr2O3@P-RuO2的EDS元素分布图显示Ru、O、P、Ni和Cr在整个纳米纤维中均匀分布(图1j)。这表明P成功掺杂到RuO2中,并且Ni和Cr2O3纳米颗粒实现了均匀锚定。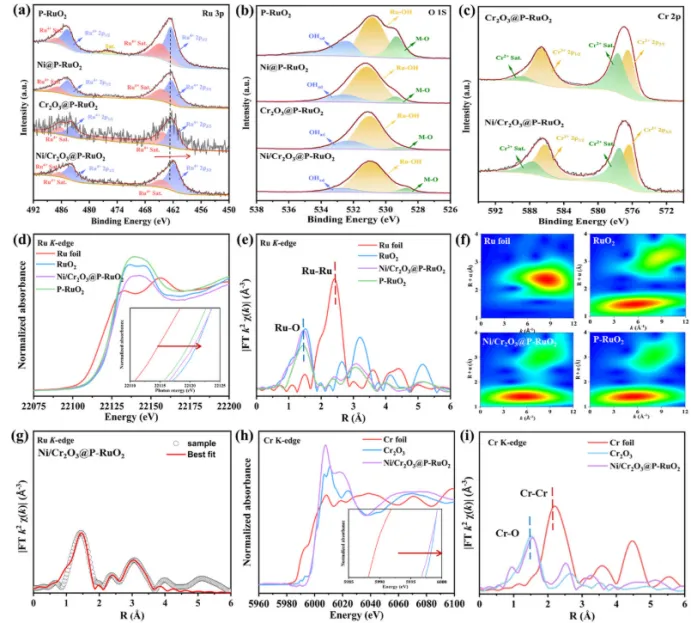
图2. 所得样品的高分辨XPS图:(a) Ru 3p, (b) O 1s, (c) Cr 2p;(d) Ru K-edge XANES谱图、(e) R空间Ru K-edge FT-EXAFS谱图、(f) Ru箔、RuO2、Ni/Cr2O3@P-RuO2气凝胶和P-RuO2纤维的WT-EXAFS等高线图、(g) Ru K-edge的R空间拟合、(h) Cr K-edge XANES谱图、(i) Cr箔、Cr2O3和Ni/Cr2O3@P-RuO2气凝胶的R空间Cr K-edge FT-EXAFS谱图。
此外,采用XPS研究了样品的表面电子态。具体而言,P-RuO2纳米纤维的Ru 3p谱图在484.6 eV和462.3 eV处呈现出两个显著峰,分别对应于Ru4+的Ru 3p1/2和Ru 3p3/2态。此外,在487.7 eV和466.1 eV处观察到Ru4+的卫星峰(图2a)。可以看出,随着Ni和Cr2O3的负载,与对照样品相比,Ru4+的结合能发生负移。这是因为Ru具有最高的电负性,导致电子从Ni和Cr向Ru迁移。在O 1s精细谱中,位于532.8、530.9和529.5 eV的峰分别对应于表面吸附的羟基物种(OHad)、Ru-OH和M-O(图2b)。值得注意的是,Ni和Cr2O3的负载降低了Ru-O键峰的面积,表明Ni和Cr2O3的引入削弱了Ru-O键的共价特性。P 2p谱图中133.3 eV和130.0 eV处的峰可分别归属于P-O键和Ru-P键。这表明P以POx单元的形式取代了RuO2晶格中的O原子,这与XRD精修结果一致。此外,Cr 2p谱图对应于Cr3+及其卫星峰(图2c)。这些结果揭示了P-RuO2与其上负载的Ni/Cr2O3界面处存在强烈的电子转移现象。
进一步采用XAS评估了Ni/Cr2O3@P-RuO2气凝胶中Ru、Ni和Cr位点的价态和配位环境。图2d展示了Ni/Cr2O3@P-RuO2气凝胶和P-RuO2的Ru K-edge XANES谱图,以Ru箔和商业RuO2作为对照。可以看出,与P-RuO2相比,Ni/Cr2O3@P-RuO2的Ru吸收边向低能方向移动,表明其平均氧化态降低。这证实了RuO2周围存在电子富集,与XPS分析结果一致。因此认为Ni/Cr2O3@P-RuO2异质界面处的电子再分布是RuO2获得电子的主要来源。为了平衡界面电化学势,电子通过内建电场从Ni/Cr2O3流向RuO2。这种热力学自发过程优化了Ru位点的电子结构,从而调节了含氧中间体的吸附强度,协同增强了OER和HER的催化活性。Ru K-edge的FT-EXAFS谱图显示在约1.50 Å处有一个主峰,对应于第一配位壳层中Ru-O键的散射路径(图2e)。我们进一步采用K空间和R空间高分辨率WT-EXAFS分析来研究原子成键。在Ni/Cr2O3@P-RuO2和P-RuO2的WT等高线图中,仅观察到一个与Ru-O信号相关的最大强度峰(图2f)。定量FT-EXAFS拟合表明,与P-RuO2相比,Ni/Cr2O3@P-RuO2中Ru-O的配位数降低,同时XANES吸收边负移(图2g)。这表明Ni/Cr2O3的引入诱导了Ru位点局部配位环境的改变,从而促进了配位不饱和位点的形成。这可能是作为亲核位点促进OER中间体优先吸附,从而降低中间体结合能、增强动力学。此外,对Ni/Cr2O3@P-RuO2中Cr K-edge和Ni K-edge的XANES分析显示,在1.50 Å处有一个特征峰,对应于Cr-O和Ni-O键(图2h,i)。在EXAFS谱图中更高R空间位置检测到了Cr-Ru和Ni-Ru的散射路径,证实负载在RuO₂表面的Ni/Cr₂O₃形成了异质界面。这种界面电荷再分布对于调控电子结构、形成配位不饱和位点以及最终增强催化电解反应活性起着关键作用。
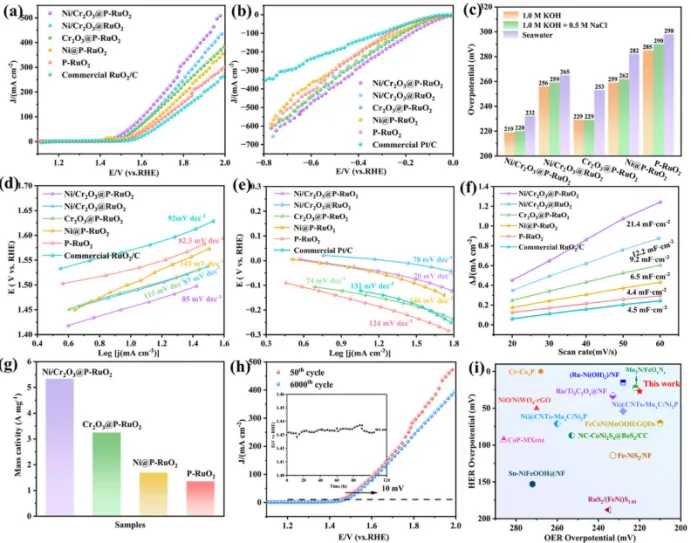
图3. (a) OER的LSV极化曲线、(b) HER的LSV极化曲线;(c) 不同电解质中OER过电位的统计直方图;(d) OER的Tafel斜率图、(e) HER的Tafel斜率图;(f) 用于ECSA计算的双电层电容与扫描速率的线性关系图;(g) 质量活性的统计直方图;(h) 6000圈CV扫描前后的LSV曲线(插图为Ni/Cr2O3@P-RuO2气凝胶在10 mA cm-2恒电流密度下持续120 h的电位曲线);(i) 所制备的Ni/Cr2O3@P-RuO2气凝胶与文献中其他已报道电催化剂的OER性能对比。
为了系统评估Ni/Cr摩尔比对催化性能的影响,制备了一系列Ni/Cr2O3@P-RuO2气凝胶样品,其Ni/Cr摩尔比呈梯度变化,同时保持总金属负载量以及与RuO2的摩尔比恒定。采用三电极体系评价了这些催化剂的HER和OER性能。LSV结果表明,Ni:Cr2O3比例为1:1的样品表现出最佳的OER和HER性能。具体而言,在1.0 M KOH溶液中,Ni/Cr2O3@P-RuO2气凝胶在10 mA cm-2电流密度下的OER和HER过电位分别低至219/25 mV。此外,在模拟海水(1.0 m KOH + 0.5 m NaCl)和碱性天然海水中,Ni/Cr2O3@P-RuO2气凝胶样品的OER和HER过电位分别为220/27 mV和232/37 mV,显著低于Ni/Cr2O3@P-RuO2、Ni@P-RuO2、Cr2O3@P-RuO2、P-RuO2以及商业Pt/C和RuO₂/C(图3a-c)。该结果表明,Cr2O3保护层的形成并未阻碍RuO2的电催化性能。同时,Ni与Cr2O3之间的协同作用,结合P掺杂策略,有效提高了P-RuO2的活性,展现出优异的HER和OER催化性能。此外,通过Tafel斜率测量评估了所制备气凝胶催化剂在1 m KOH + 0.5 m NaCl溶液中OER和HER过程的反应动力学。Ni/Cr2O3@P-RuO2的OER Tafel斜率为87 mVdec-1,HER Tafel斜率为70 mVdec-1,显著低于其他对照样品(图3d,e)。这些结果表明,Ni/Cr2O3@P-RuO2的多元素协同效应诱导的电子结构调控增强了活性位点的本征活性,从而在催化过程中表现出优异的反应动力学特性。为了进一步阐明Ni/Cr2O3@P-RuO2的催化反应活性,该催化剂表现出最高的Cdl为21.4 mFcm-2,显著高于其他对照样品和商业RuO2/C。这表明Ni/Cr2O3@P-RuO2具有更大的ECSA,从而暴露出更多的活性位点(图3f)。此外,质量活性(MA)是量化单位质量电化学活性的重要指标,直接反映了电催化剂可用活性位点的密度。在300 mV过电位下,基于ICP-OES结果的MA表明,Ni/Cr2O3@P-RuO2气凝胶催化剂表现出优异的本征活性(5.32 Amg-1)(图3g)。稳定性测试进一步证实了所制备催化剂的实际应用潜力。在6000次连续CV循环后,Ni/Cr2O3@P-RuO2的过电位仅衰减10 mV(图3h)。重要的是,与先前报道的电催化剂相比,Ni/Cr2O3@P-RuO2气凝胶仍然表现出出色的OER性能(图3i)。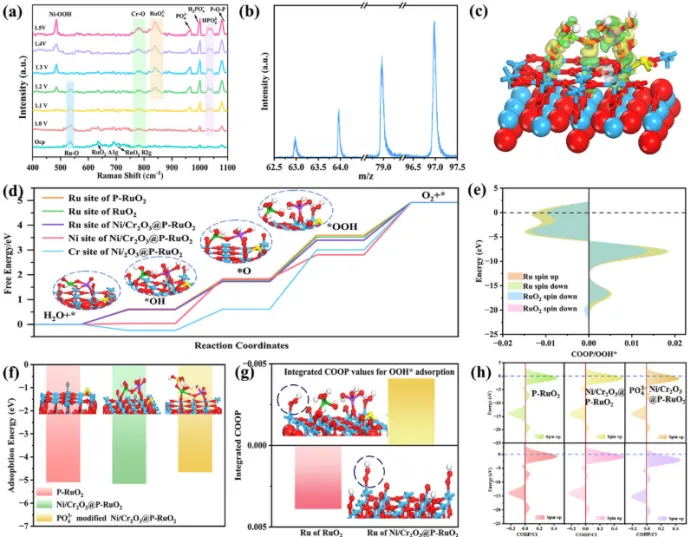
图4. (a) Ni/Cr2O3@P-RuO2在不同外加电位下用于OER的原位拉曼光谱;(b) OER后Ni/Cr2O3@P-RuO2气凝胶的TOF-SIMS谱图;(c) 优化后的*OOH吸附在Ni/Cr2O3@P-RuO2模型上的电子密度差图(黄色代表得电子,绿色代表失电子);(d) 在U = 1.23 V下四个OER基元步骤吉布斯自由能曲线的DFT计算结果(白色、红色、蓝色、绿色、紫色、黄色和灰色原子分别代表H、O、Ru、Ni、Cr、P和Cl原子);(e) Ru与*OOH之间键的COOP图;(f) *Cl吸附的自由能计算及原子结构图;(g) 计算得到的COOP值;(h) Ru位点与Cl-之间键的COOP图。
为进一步揭示OER过程中催化剂的表面重构情况,在1.0至1.5 V(相对RHE)的电位范围内记录了电化学原位拉曼光谱(图4a)。在~530 cm-1处观测到Eg带,~630 cm-1处的A1g带以及~688 cm-1处的B2g带,它们分别对应于RuO6八面体的弯曲振动、对称伸缩振动和反对称伸缩振动,证实样品保持金红石结构。随着电位升高,可以观测到归属于Ni-O键弯曲振动的峰,这表明原位生成了具有高OER活性的高价态Ni。这是因为高价金属离子提供更多的未占据d轨道,与中间体的p轨道形成更强的相互作用,从而促进吸附能并提高整体催化活性。此外,~781 cm-1附近的宽峰对应于Cr-O键。值得注意的是,在~840 cm-1附近观测到归属于RuO42-的峰,进一步证实了在OER过程中Ru4+被氧化为Ru6+,且没有可溶性的Ru8+存在。另外,~965、1001、1079和1037 cm-1附近的峰分别对应于PO43-、H2PO4-、P-O-P和HPO42-,这表明在OER过程中生成了一系列含磷阴离子物种。为了进一步分析PO43-的原位形成,采用了飞行时间二次离子质谱(TOF-SIMS)的负离子模式进行分析(图4b)。可以看出,在质荷比(m/z)为62.9、63.9、78.9和96.9处显示的特征碎片峰分别对应于PO2-、HPO2-、PO2-和H2PO4-。这表明掺入RuO2晶格中的P在OER的高氧化电位下发生P-O-Ru键断裂、溶出和重构,最终以H2PO4-/PO43-的形式富集在催化剂表面。这种带负电的保护层能够有效排斥Cl-,显著增强催化剂的耐腐蚀性能。
随后利用DFT计算系统研究了Ni/Cr2O3@P-RuO2催化活性提升的机理。基于电化学原位拉曼光谱的结果,建立了将NiCr羟基氧化物簇锚定在P-RuO2末端O上的几何优化构型。Ni/Cr2O3@P-RuO2优化构型的电子密度差和电子密度切片显示,与Ni和Cr原子周围区域相比,Ru原子周围存在电子聚集,表明电子从Ni/Cr2O3流向RuO2(图4c)。OER中的速率决定步骤(RDS)对应于自由能变化中热力学势垒最高的阶段。如图4d所示,在所有潜在活性位点中,RuO2和P-RuO2的Ru位点以及最优Ni/Cr2O3@P-RuO2的Ru和Cr位点,在*O向*OOH转化时均表现出最大的吉布斯自由能变化(ΔG3),这可视为整个OER过程的RDS。这些位点的ΔG3表明,Ni/Cr2O3@P-RuO2的Ru位点(1.66 eV)的值显著低于RuO2的Ru位点(1.76 eV)、P-RuO2的Ru位点(1.79 eV)以及Ni/Cr2O3@P-RuO2的Cr位点(2.39 eV)。此外,Ni位点的RDS是第二步反应(ΔG2 = 1.80 eV),其*O的结合能明显强于Ni/Cr2O3@P-RuO2的Ru位点(ΔG2 = 1.13 eV)。这些结果表明,Ni/Cr2O3@P-RuO2的Ru位点对*O的吸附能处于适中水平,既不强也不弱,因此该Ru位点被认为是OER过程中最可能的活性位点。另外,为了阐明Ni/Cr2O3@P-RuO2电催化剂的HER机理,我们评估了氢吸附自由能(ΔG*H),这是基于Sabatier原理的一个关键热力学描述符。晶体轨道重叠布居(COOP)分析进一步阐明了Ru原子与*OOH中O原子之间的成键和反键相互作用。COOP分析表明,在-12.08 eV至-5.83 eV的能量范围内,相互作用主要为成键,而在-5.83 eV至费米能级(0 eV)之间出现了明显的反键态(图4e)。为了定量比较吸附强度,我们计算了整个能量区间内的总COOP积分值。结果表明,Ni/Cr2O3@P-RuO2的Ru位点与*OOH之间键的COOP积分值为0.0039,呈现成键效应。而RuO₂的Ru位点与*OOH之间键的COOP积分值为-0.0041,呈现反键效应。这直接证实了Ni/Cr2O3@P-RuO2的Ru位点对*OOH具有更强的吸附能力,从而有助于加速OER反应动力学。基于电化学原位拉曼光谱表明P溶解并氧化成PO43-物种的结果,我们计算了Cl-在PO43-修饰的Ni/Cr2O3@P-RuO2上吸附于Ru位点的吸附能,以进一步阐明耐腐蚀机理(图4g)。结果表明,PO43-修饰的Ni/Cr2O3@P-RuO2对Cl-的吸附能ΔECl-(-0.46)相比于Ni/Cr2O3@P-RuO2(-0.52)和P-RuO2(-0.51)更正。这表明PO43-的引入显著削弱了Cl-在催化剂表面的吸附。为了在电子结构水平上理解这一现象,我们进一步计算了活性位点Ru与吸附Cl-之间的COOP(图4f)。计算结果表明,PO43-修饰的Ni/Cr2O3@P-RuO2的COOP值(0.029)明显低于Ni/Cr2O3@P-RuO2(0.033)和P-RuO2(0.035),表明PO43-的引入导致Ru与Cl-之间的成键最弱(图4g,h)。因此,这些结果共同证实了原位形成的PO43-在增强催化剂整体抗ClER能力方面发挥着重要作用。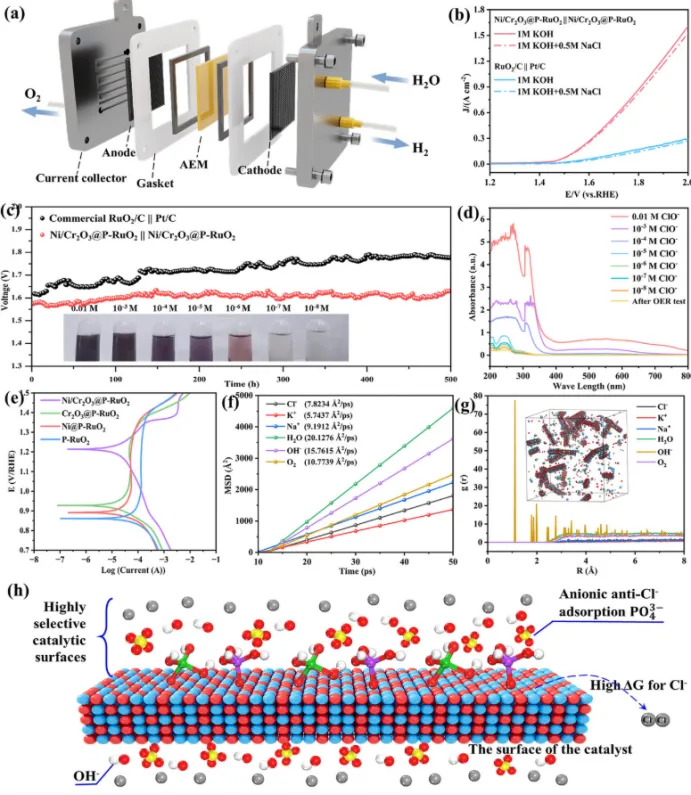
图5. (a) AEM电解槽示意图;(b) AEM电解槽的LSV曲线;(c)Ni/Cr2O3@P-RuO2催化下AEMSE电解槽在100 mAcm-2下的耐久性测试;(d) 电解质中次氯酸根物种的紫外-可见吸收光谱;(e) 所得样品的动电位极化曲线;(f) MSD对比图;(g) RDF图像;(h) concise电催化示意图。
所制备的Ni/Cr2O3@P-RuO2同时作为阳极和阴极,组装到AEMWE和AEMSE系统中,并在70°C下评估其工业应用性能(图5a)。三种阴离子交换膜(Fumasep FAA-3-50、FAA-3-PK-75和FAA-3-PK-130)被分别组装到AEMWE和AEMSE中,以评估它们的水电解性能。其中,FAA-3-PK-130在腐蚀性环境中表现出更高的机械稳定性,因此在测试中被用作电解池中的AEM。Ni/Cr2O3@P-RuO2在1 M KOH + 0.5 M NaCl中达到1.5 Acm-2电流密度时的槽压为1.9 V,在碱化天然海水中达到1.5 Acm-2时的槽压为2 V,显著优于商业Pt/C和RuO2/C体系(图5b)。得益于Ni/Cr2O3@P-RuO2气凝胶催化剂构建的Cr2O3层与原位生成阴离子所形成的双重抗腐蚀保护机制,AEMSE在100 mAcm-2下连续运行500小时后,电压衰减仅为60 µVh-1(图5c),而商业Pt/C和RuO2/C催化剂在短时间内即表现出显著的电流衰减。这些结果表明,与对照样品相比,它表现出更优异的耐腐蚀性能。为了研究海水中杂质离子对催化剂的影响,采用分子动力学模拟计算了催化剂表面的扩散行为和局部密度演化。模拟过程中的构型如图5e所示。均方位移(MSD)分析表明,H2O和OH-具有更高的扩散速率,而Cl-和K+的扩散能力显著较低(图5f)。这表明在该体系中,OH-和H2O具有更优的迁移能力,能够优先占据催化剂表面的活性位点,从而有效抑制Cl-等杂质离子的吸附,显著增强催化剂的抗Cl-腐蚀能力。径向分布函数(RDF)显示,在催化剂表面约8 Å范围内存在显著的OH-富集(图5g)。其第一配位层的峰强度明显高于其他离子,表明OH-对催化剂表面具有很强的亲和力。相比之下,Cl-等杂质离子在该区域的分布浓度极低,没有明显的配位峰。该结果证实催化剂表面对OH-具有优先吸附性,形成了局部高OH-浓度的微环境,通过静电排斥有效阻挡了Cl-的吸附。综上所述,Ni/Cr2O3@P-RuO2通过两种协同机制抵抗腐蚀:Cr2O3作为路易斯酸选择性吸附OH-,以及P掺杂原位生成的PO43-通过静电相互作用排斥Cl-。因此,这种协同双机制为增强海水电解中催化剂的耐腐蚀性能提供了一种有效的策略。5、总结与展望
总之,该研究报道了一种由锚定在P-RuO2纳米纤维上的Ni/Cr2O3纳米颗粒组成的三维多孔气凝胶,用于海水电解催化。该Ni/Cr2O3@P-RuO2气凝胶在模拟海水中对OER和HER分别表现出220/27 mV@10 mA cm-2的低过电位,并且在组装成AEMSE后能够在0.1 A cm-2下稳定运行500小时。XANES分析结合DFT计算共同揭示,多金属协同效应诱导了电子从Ni/Cr2O3向Ru转移。此外,Ru作为催化活性位点表现出不饱和配位缺陷,从而增强了OER/HER活性。原位电化学拉曼光谱和TOF-SIMS表明,原位生成的PO43-通过静电作用排斥Cl-,形成优先富集OH-的表面微环境。同时,分子动力学模拟证实了这种涉及原位生成PO43-和Cr2O3保护层的双重抗腐蚀机制的建立,显著增强了抗Cl-能力,从而克服了海水电解中的耐久性瓶颈。
6、文章链接
《Dual Anti-Corrosion Mechanisms Enabled by Lewis Acid and PO43− Modification for Alkaline Seawater Electrolysis With Ni/Cr2O3 Anchored on P-RuO2 Fibrous Aerogel》
https://doi.org/10.1002/adfm.75248


如需转载或投稿请联系我们:energy_catalysis@163.com