南京鼓楼医院 | 突破!Midkine-LRP1 轴介导肾损伤后认知损伤关键通路
- 2026-06-02 00:02:53

导读
本研究以单侧肾缺血再灌注损伤小鼠模型为基础,围绕急性肾损伤(AKI)引发认知障碍的机制展开,先验证肾损伤可导致小鼠空间学习记忆受损、血脑屏障破坏与海马神经炎症,再通过单细胞测序与配体 - 受体互作分析,锁定肾损伤高表达的 Midkine(MDK)及其受体 LRP1 为核心调控轴,证实肾损伤后肾小管细胞与成纤维细胞大量分泌 MDK,经血液循环进入海马,通过 LRP1 被小胶质细胞内吞,上调 P2ry12 表达,促使小胶质细胞向促炎 M1 型极化、吞噬功能异常增强,引发神经炎症与神经元损伤,最终导致认知障碍;抑制肾组织 MDK 表达可显著改善认知损伤、减轻小胶质细胞活化,明确 MDK-LRP1 轴是肾损伤介导认知障碍的关键通路与潜在治疗靶点。
注:该文章发表于《Advanced Science》,最新影响因子为14.1,位列JCR和中科院分区Q1/1区。
研究亮点
首次揭示MDK-LRP1-P2ry12轴介导肾 - 脑跨器官通讯,阐明缺血肾损伤通过分泌 MDK 激活海马小胶质细胞引发认知障碍的全新机制;明确肾源性 MDK 经血液循环作用于脑,为 AKI 相关认知障碍提供首个可靶向的分子通路。
研究背景
急性肾损伤(AKI)住院患者发生率约 13.3%,易进展为慢性肾病,30%-60% 的 AKI 患者会出现认知障碍,且肾功能越差认知损伤越重,传统观点认为尿毒症毒素蓄积是主因,但肾损伤远程器官调控机制尚未明确。Midkine(MDK)在肾缺血 / 毒性损伤后显著上调,可作为肾损伤标志物,同时参与中枢神经系统炎症与认知损伤调控;小胶质细胞作为脑内固有免疫细胞,异常活化会引发过度突触修剪导致认知下降,但其在 AKI 相关认知障碍中的作用及上游调控因子尚不清晰。基于此,本研究聚焦肾损伤分泌因子对海马小胶质细胞的远程调控,探索 AKI 引发认知障碍的跨器官分子机制,为临床干预提供理论依据。
研究方法
构建小鼠单侧肾缺血再灌注损伤(IRI)模型,设置假手术与不同时间点损伤组,采用 Morris 水迷宫检测认知功能,伊文思蓝染色、S100β 与紧密连接蛋白检测评估血脑屏障完整性;对肾与海马组织进行单细胞 RNA 测序,结合 CellChat 分析配体 - 受体互作;运用 qPCR、Western blot、免疫荧光检测 MDK、LRP1、P2ry12 及炎症因子表达;通过流式细胞术分析小胶质细胞极化与数量变化;构建 MDK 过表达细胞共培养体系、LRP1 敲低细胞模型,体外验证 MDK 内吞与小胶质细胞活化机制;经肾局部注射 MDK-shRNA 腺病毒,在体内验证抑制 MDK 的干预效果。
研究结果
小鼠肾 IRI 后出现明显认知障碍、血脑屏障破坏、海马神经元损伤与神经炎症;单细胞测序显示肾损伤后 MDK-LRP1 配体 - 受体互作显著增强,MDK 主要由损伤肾小管细胞与成纤维细胞分泌,经血液富集于海马,海马自身不合成 MDK;MDK 通过 LRP1 介导小胶质细胞内吞,剂量与时间依赖性上调 P2ry12 表达,促使小胶质细胞向 M1 型极化、吞噬功能异常增强,引发持续神经炎症;敲低肾组织 MDK 可降低血清与海马 MDK 水平,显著改善小鼠认知功能,减轻海马神经元凋亡、小胶质细胞活化及炎症因子释放,证实 MDK 是介导肾损伤后认知障碍的关键因子。
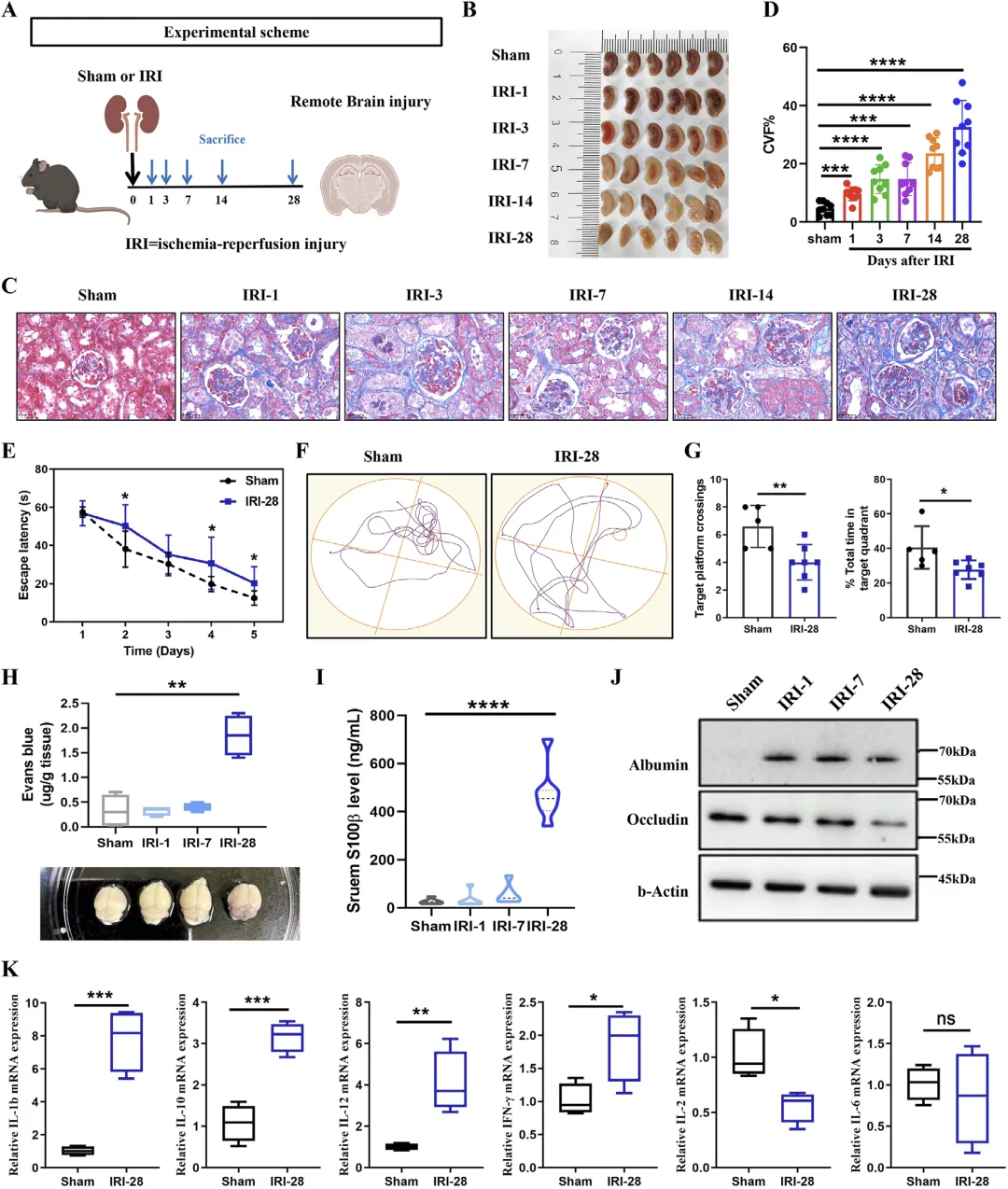
Figure 1 肾缺血损伤对认知功能、血脑屏障及组织炎症的系统性影响
这张图全面证明小鼠单侧肾缺血再灌注损伤后,会从肾脏损伤逐步发展为慢性纤维化,同时引发明确的认知障碍、血脑屏障破坏与海马神经炎症,完整建立起 “急性肾损伤→慢性肾病→远程脑损伤” 的动物模型。实验按时间梯度观察肾脏损伤进程,组织形态与染色结果显示损伤后肾小管坏死、萎缩并逐渐出现间质纤维化,肾功能指标显著异常;Morris 水迷宫实验直接证实损伤小鼠空间学习与记忆能力明显下降,寻找平台时间延长、目标象限停留与穿越次数减少;伊文思蓝渗漏、血清 S100β 升高、紧密连接蛋白 Occludin 降低等结果共同说明血脑屏障完整性被破坏;海马组织出现神经元形态损伤、核固缩,多种促炎因子表达显著上调,充分说明肾缺血损伤不仅造成局部肾脏病变,还通过全身途径引发脑部炎症、神经元损伤与认知功能下降,为后续机制探索提供可靠模型依据。
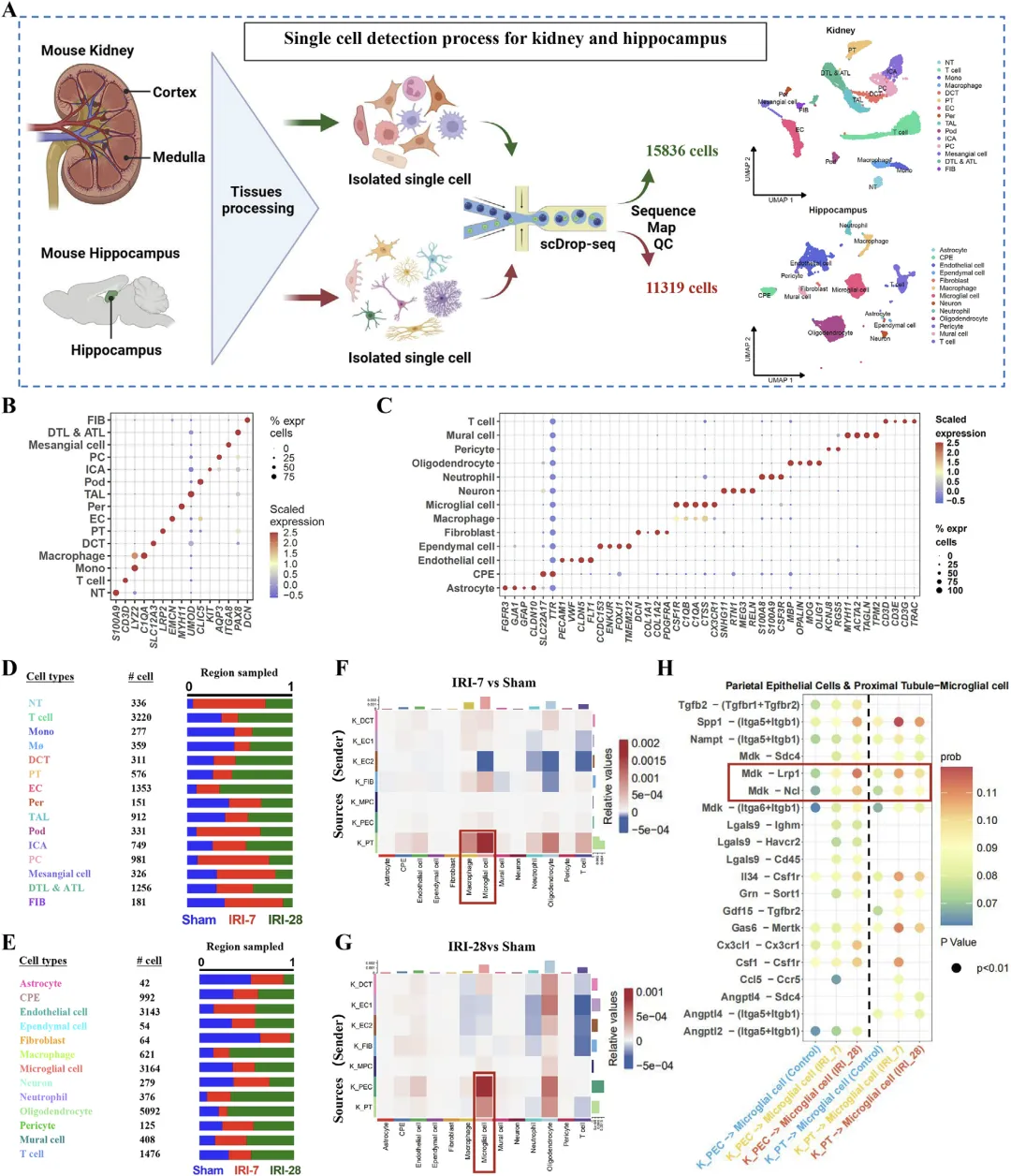
Figure 2 单细胞测序揭示肾损伤后肾 - 海马跨器官细胞通讯与分子网络重塑
这张图以单细胞 RNA 测序为核心手段,系统解析肾缺血损伤后肾脏与海马组织的细胞类型变化与跨器官分子对话,锁定介导肾 - 脑通讯的关键配体 - 受体通路。研究分别对肾脏与海马组织进行单细胞测序,质控后获得大量有效细胞数据,分群注释出两类器官的主要细胞亚型;通过 CellChat 配体 - 受体互作分析,对比假手术组、损伤早期与晚期样本,发现肾损伤后肾脏近端小管等细胞与海马小胶质细胞之间的细胞通讯强度显著增强;在众多信号轴中,MDK-LRP1 配对的相互作用信号最为突出且具有统计学意义,明确这条通路是肾损伤后介导肾脏向海马传递信号、调控小胶质细胞的核心候选通路,为后续聚焦 MDK-LRP1 轴开展机制研究提供直接的单细胞水平证据。
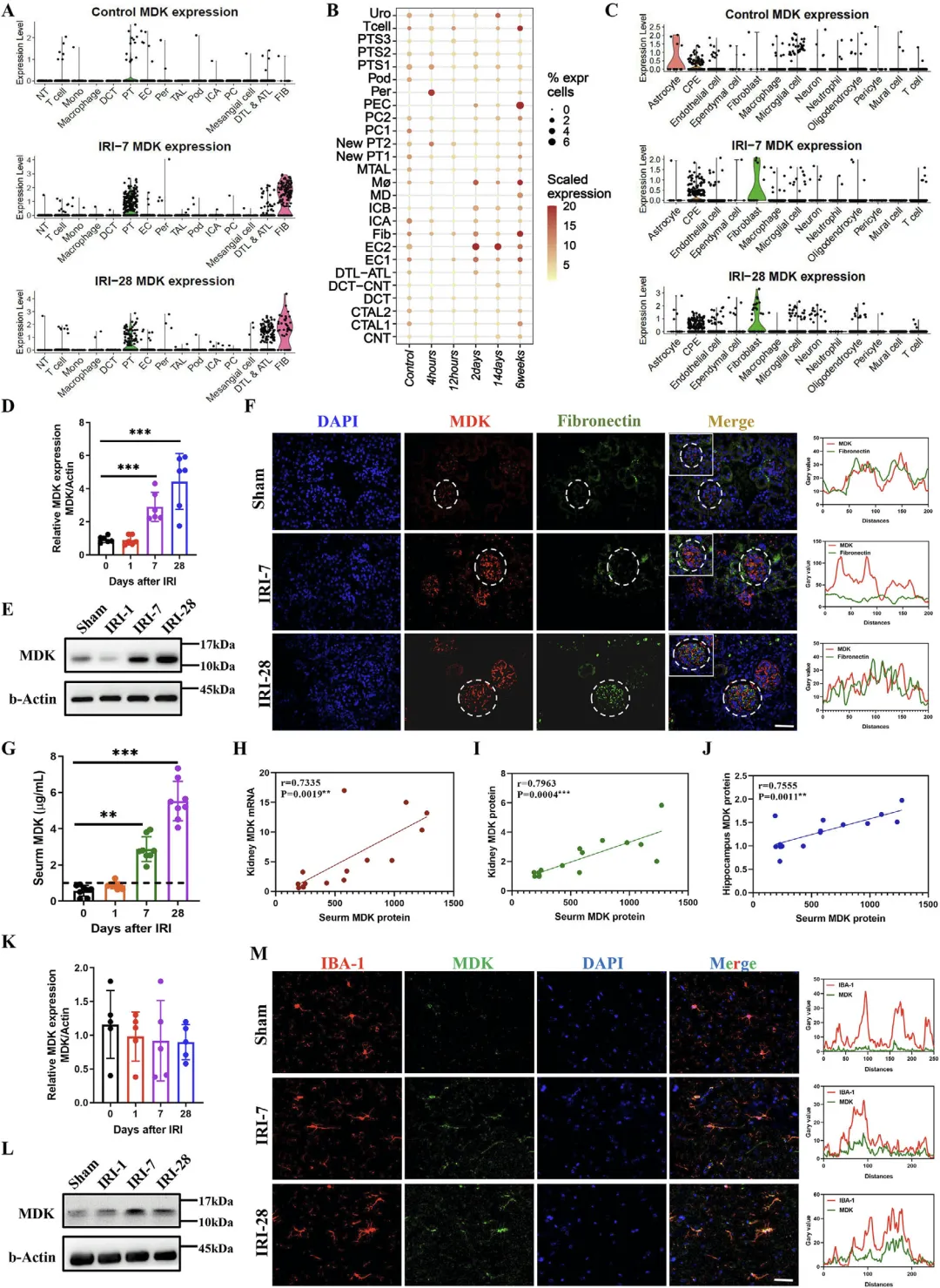
Figure 3 肾缺血损伤后肾脏源 MDK 高表达并经循环富集于海马组织
这张图系统阐明 Midkine(MDK)在肾损伤后的表达来源、动态变化与组织分布规律,证实海马中升高的 MDK 主要来自损伤肾脏而非脑部自身合成。单细胞测序显示正常肾脏几乎不表达 MDK,损伤后肾小管细胞与成纤维细胞显著高表达 MDK,并随损伤进程持续升高,公共数据集验证结果与本研究一致;qPCR 与 Western blot 从 mRNA 和蛋白水平进一步证实肾脏 MDK 随损伤时间上调,免疫荧光显示 MDK 主要定位在肾小管与成纤维细胞;血清 MDK 水平与肾脏 MDK 表达呈显著正相关,同时海马组织中 MDK 蛋白也随损伤时间逐渐升高,但海马单细胞测序显示其自身几乎不合成 MDK,免疫荧光显示 MDK 与小胶质细胞标记物 IBA-1 共定位,充分证明损伤肾脏大量分泌 MDK 进入血液循环,透过受损血脑屏障在海马富集并靶向小胶质细胞。
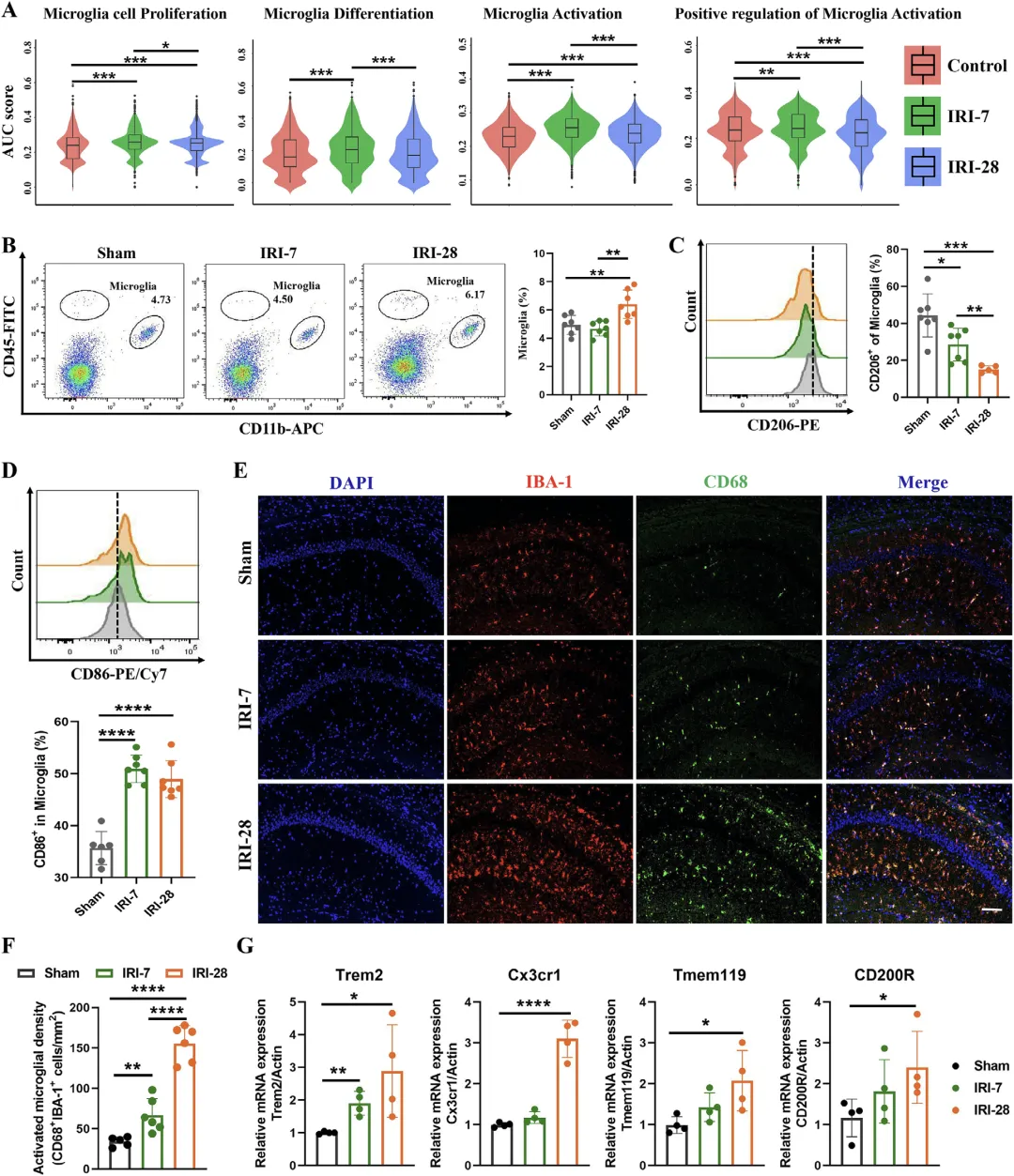
Figure 4 缺血性肾损伤诱导海马小胶质细胞持续活化并呈促炎表型
这张图从单细胞、细胞形态与分子表达多个层面,证实肾缺血损伤可引发海马小胶质细胞大量增殖、异常活化并向促炎 M1 型极化,形成持续神经炎症状态。单细胞测序的小提琴图显示损伤后小胶质细胞的增殖、分化与活化通路显著激活;流式细胞术定量检测发现损伤小鼠海马中小胶质细胞数量明显增多,促炎 M1 表型标记 CD86 比例大幅上升,抗炎 M2 表型 CD206 比例下降,呈现稳定促炎极化;免疫荧光直观显示活化小胶质细胞数量与 CD86 荧光强度显著升高;同时小胶质细胞活化相关标记基因 Trem2、Cx3cr1、Tmem119 等表达显著上调,共同说明肾损伤通过远程信号驱动海马小胶质细胞过度活化,形成促炎微环境,为后续认知障碍提供细胞学基础。
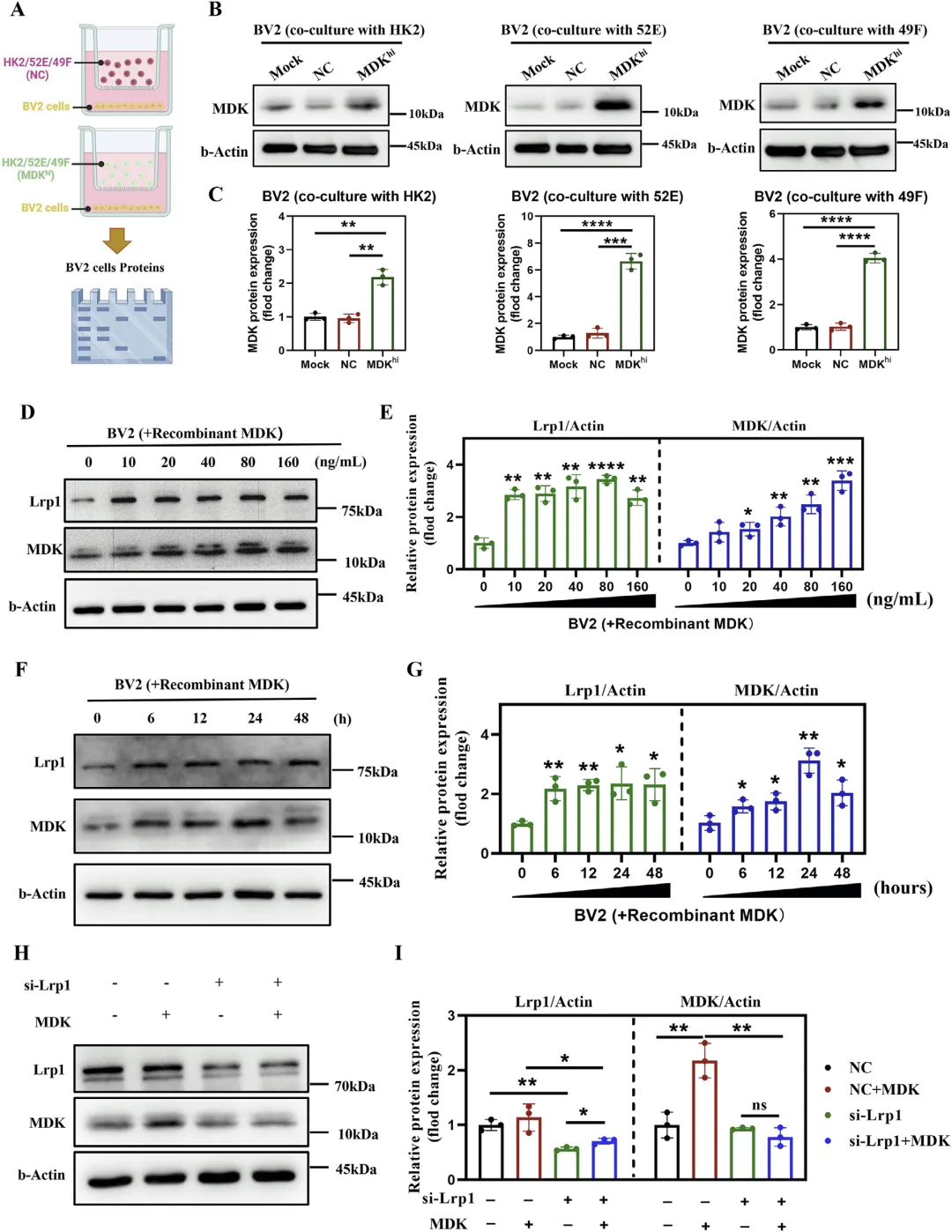
Figure 5 MDK 通过 LRP1 受体介导被小胶质细胞内吞的分子机制
这张图通过体外细胞实验严格证实,肾脏来源的 MDK 必须依靠 LRP1 受体才能进入小胶质细胞,是 MDK 发挥作用的关键入口。研究构建 MDK 过表达的肾小管上皮细胞与成纤维细胞,将其培养上清处理小胶质细胞,发现小胶质细胞内 MDK 含量显著上升;使用重组 MDK 蛋白直接处理小胶质细胞,证实 MDK 内吞呈时间与剂量依赖性,同时小胶质细胞膜 LRP1 表达适应性升高;采用 CRISPR/Cas9 或 siRNA 敲低 LRP1 后,MDK 几乎无法进入小胶质细胞,内吞过程被完全阻断;同时排除整合素 ItgA6、Itgb1 等其他 MDK 受体的参与,明确 LRP1 是介导 MDK 进入小胶质细胞的唯一关键受体,建立 “MDK→LRP1→小胶质细胞内吞” 的核心通路。
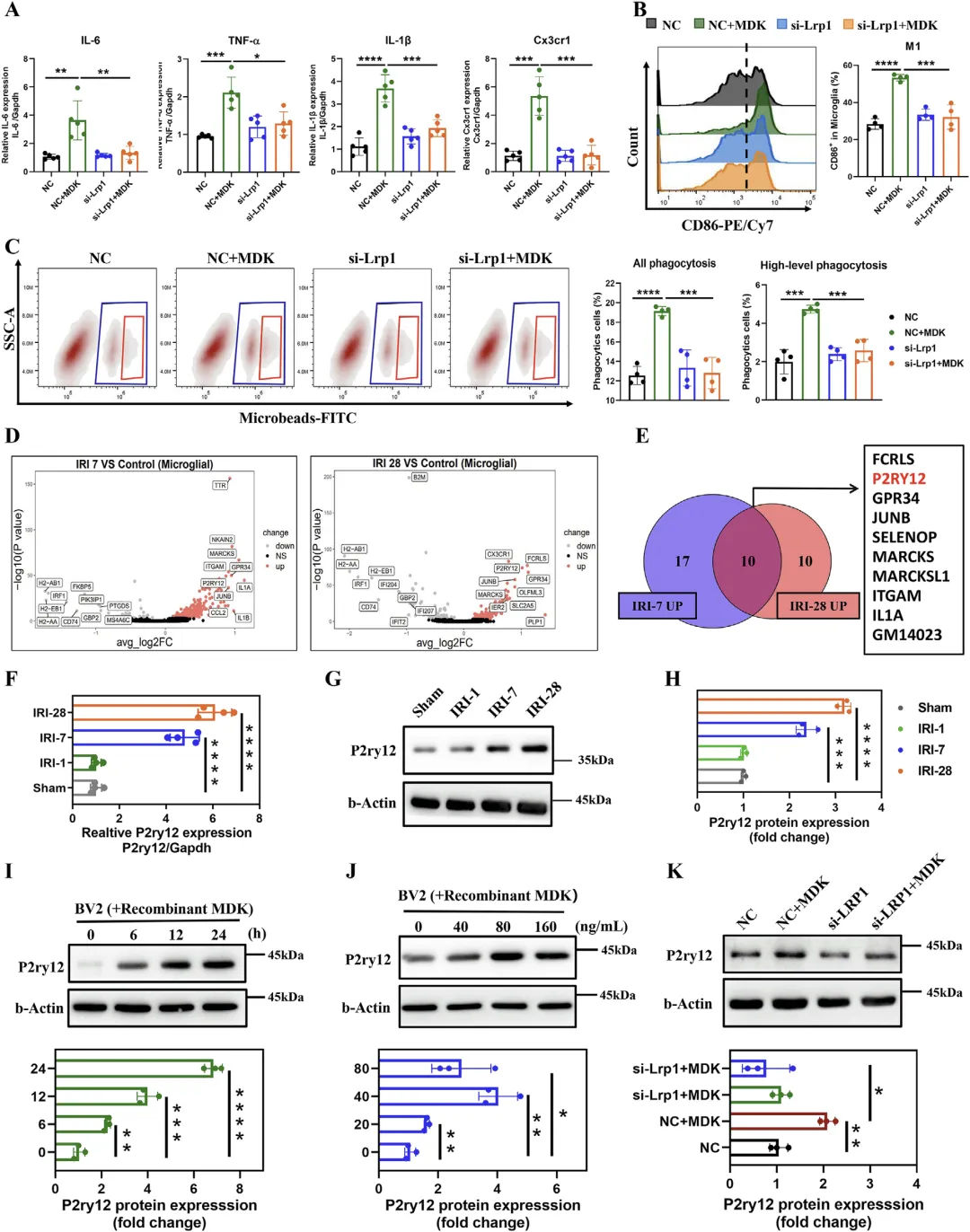
Figure 6 MDK-LRP1 轴通过上调 P2ry12 驱动小胶质细胞活化与过度吞噬
这张图深入揭示 MDK 进入小胶质细胞后引发活化的下游分子机制,证实 LRP1 介导的 MDK 内吞通过上调 P2ry12 实现促炎与功能激活。体外实验显示 MDK 处理可显著上调小胶质细胞促炎因子 IL-6、TNF-α、IL-1β 等表达,增强细胞吞噬能力,促使其向 M1 型极化;敲低 LRP1 后,上述炎症激活与吞噬增强效应完全消失,说明依赖 LRP1;单细胞测序与分子验证发现,肾损伤后海马小胶质细胞中 P2ry12 显著上调,且 MDK 以时间与剂量依赖方式上调 P2ry12,敲低 LRP1 可阻断该上调作用,明确 P2ry12 是 MDK-LRP1 下游调控小胶质细胞活化的关键效应分子,完整解析 MDK 诱导小胶质细胞功能异常的分子通路。
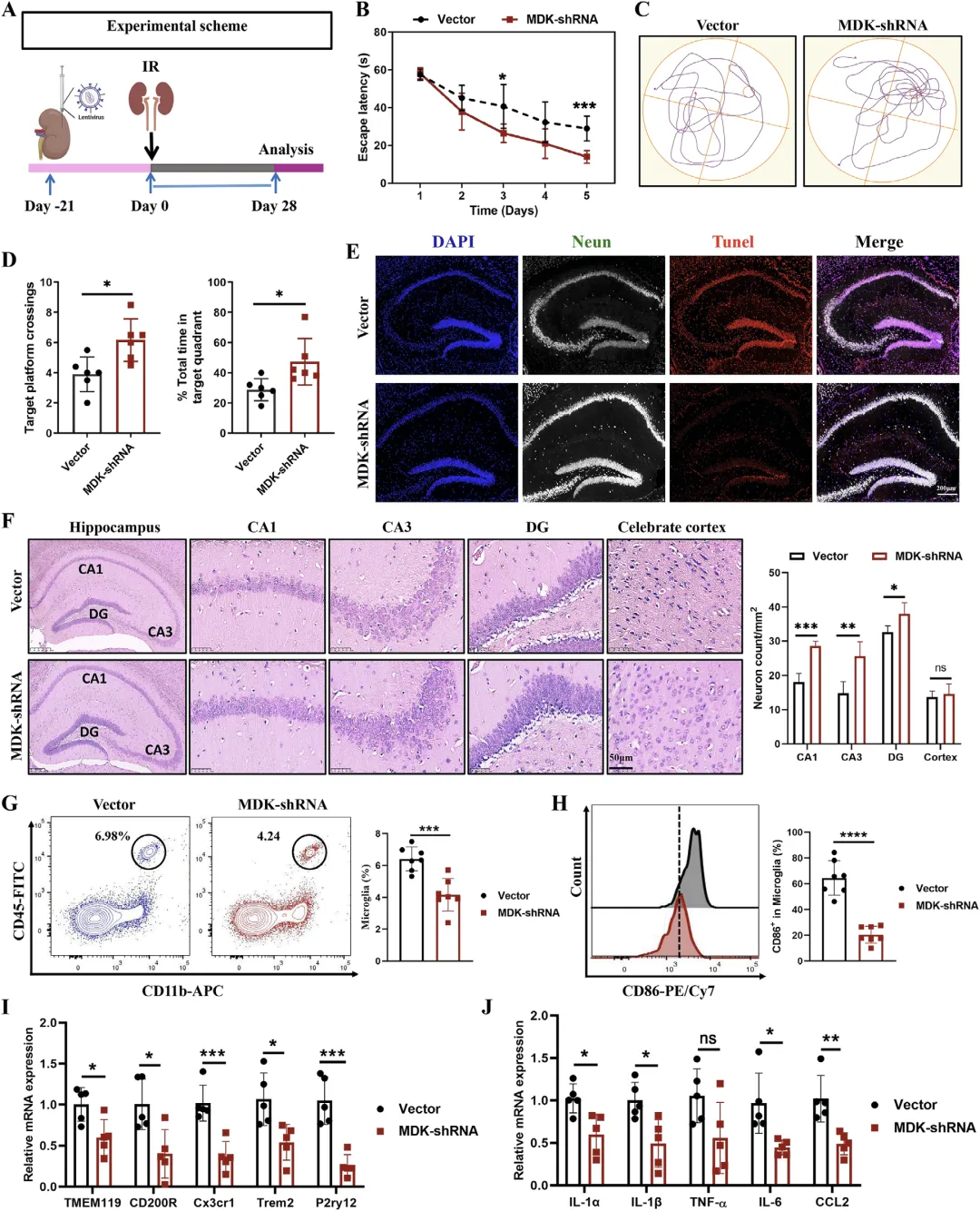
Figure 7 抑制肾脏 MDK 表达可缓解肾损伤诱导的认知障碍与小胶质细胞活化
这张图通过体内干预实验反向验证 MDK 是肾损伤引发认知障碍的关键靶点,证明抑制肾脏 MDK 表达可有效改善脑损伤与认知功能。研究在小鼠肾脏局部注射 MDK-shRNA 腺病毒特异性敲低肾脏 MDK,成功降低肾脏、血清与海马 MDK 水平;Morris 水迷宫结果显示,敲低 MDK 后肾损伤小鼠的逃避潜伏期显著缩短,平台穿越次数与目标象限停留时间明显回升,认知功能大幅改善;同时海马神经元凋亡减少、CA1/CA3/DG 区神经元损伤减轻;流式与分子检测显示小胶质细胞数量减少、M1 型活化被抑制,小胶质细胞活化标记与促炎因子表达显著下降,充分证实肾脏来源 MDK 是介导肾损伤后认知障碍的关键因子,靶向抑制肾脏 MDK 可同时缓解神经炎症与认知损伤,具备明确转化价值。
研究结论
本研究证实缺血性肾损伤通过MDK-LRP1-P2ry12轴引发海马小胶质细胞异常活化,进而导致认知障碍,MDK 主要来源于损伤肾脏,经血液循环跨越受损血脑屏障作用于海马小胶质细胞;MDK 经 LRP1 内吞后上调 P2ry12,驱动小胶质细胞促炎极化与过度吞噬,诱发神经炎症与神经元损伤,最终造成认知功能下降;抑制肾脏 MDK 表达可有效缓解肾损伤后继发的认知障碍与神经炎症,提示 MDK-LRP1 轴是治疗 AKI 相关认知障碍的潜在靶点,为肾 - 脑交互疾病提供新的治疗方向。
仅在小鼠模型验证,未开展临床样本验证;未区分海马星形胶质细胞源性 MDK 的贡献;未来需开展临床研究验证 MDK 作为标志物与靶点的价值,构建星形胶质细胞特异性敲除模型,明确中枢与外周 MDK 的协同作用。
参考文献

国家杰青一对一答疑视频
医学省自然申请答疑,立项的关键条件是哪一些?从哪些方向可以杀出重围
临床型博士如何准备国青标书?没有预实验怎么办?专家一对一解答规划
中医药科研研究