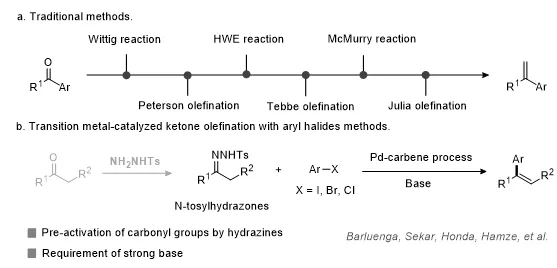
芳基取代烯烃广泛存在于药物分子、天然产物和功能材料中,是有机合成中一类重要的结构单元和转化平台。由于芳基烯烃骨架兼具良好的结构可调性和后续衍生化能力,其高效构建一直是有机合成化学中的重要研究方向。酮类化合物来源丰富、结构多样,是合成烯烃类化合物的理想前体。传统酮烯烃化方法(图1a),如Wittig反应、Horner–Wadsworth–Emmons反应、Peterson反应、Tebbe反应、McMurry反应和Julia反应等,已被广泛用于构建芳基取代烯烃。然而,这些方法通常需要预先制备特定的烯烃化试剂,且常伴随当量副产物生成、原子经济性不足、官能团兼容性受限等问题。
近年来,过渡金属催化策略为酮类化合物的烯烃化提供了新的思路。其中,酮与芳基卤化物的交叉偶联反应在实现烯化方面展现出了巨大的潜力,因为该策略可直接利用易得的羰基化合物和芳基卤化物构建结构多样的芳基烯烃。此前,相关研究主要依赖钯催化的N-磺酰腙与芳基卤化物偶联过程,即先将酮转化为腙类中间体,再通过钯卡宾途径生成烯烃。尽管该策略具有重要意义,但酮的预活化、强碱条件以及操作步骤增加等问题仍限制了其进一步应用(图1b)。因此,发展无需预活化、可直接利用酮类化合物与芳基卤化物实现烯烃构建的新方法具有重要研究价值。
图1. 研究背景。图片来源:Chin. Chem. Lett.
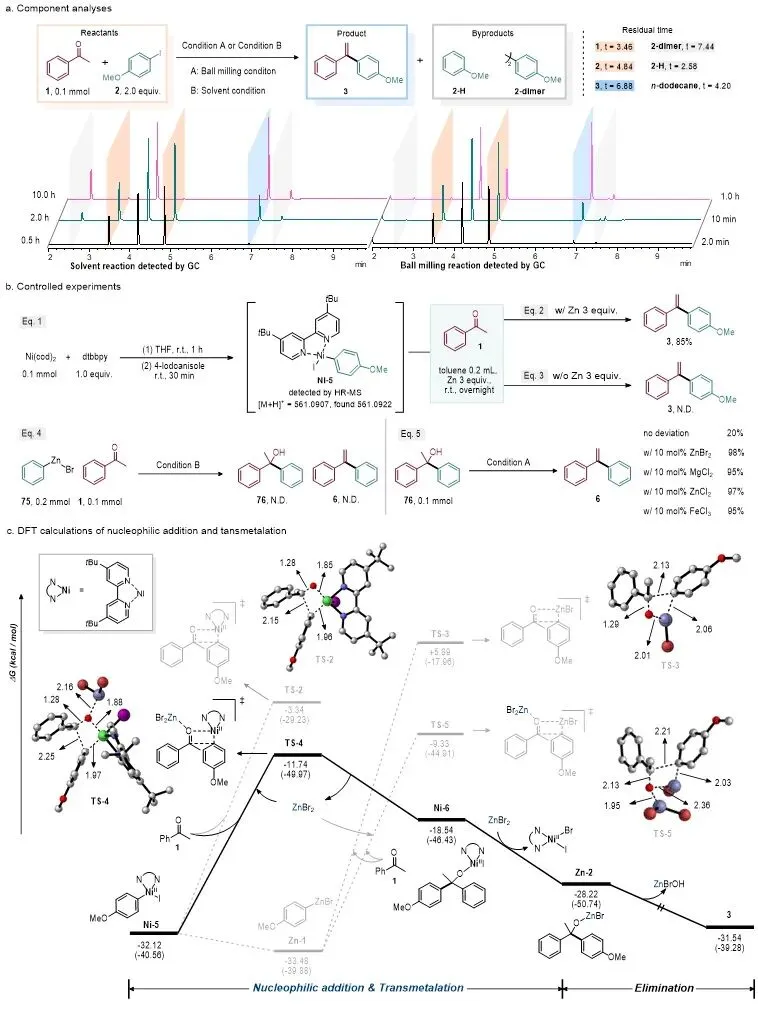
在此背景下,南京师范大学杜炳南教授团队与南京大学王毅教授团队合作,发展了一种机械化学球磨促进的镍催化酮类化合物与芳基卤化物直接还原烯烃化反应。该方法以商业易得的酮和芳基卤化物为原料,在少量THF作为液体辅助研磨剂的条件下,使用NiBr2•dtbbpy作为催化剂、锌粉作为还原剂,于室温球磨条件下即可高效生成芳基取代烯烃。该研究成果近日被Chinese Chemical Letters接收发表。
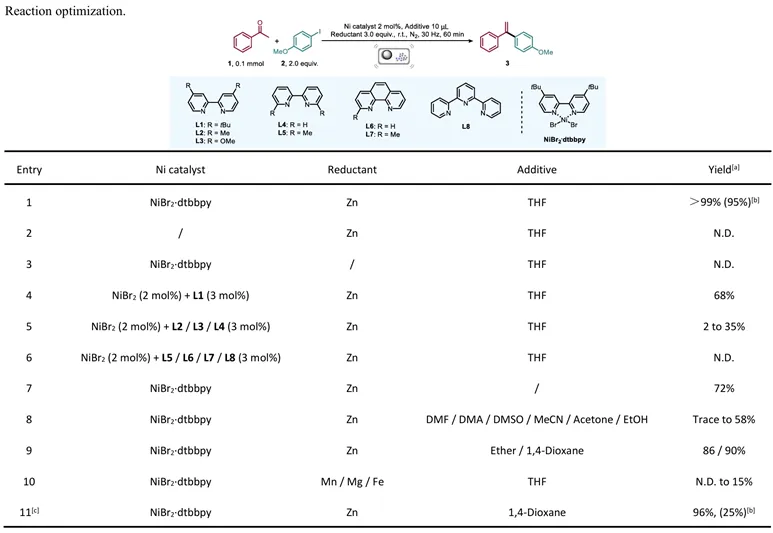
研究人员首先以苯乙酮和4-碘苯甲醚作为模型底物,对反应条件进行了系统优化(图 2)。结果表明,在30 Hz球磨条件下,以2 mol% NiBr2•dtbbpy为催化剂、3当量锌粉为还原剂,并加入10 μL THF作为添加剂,反应60分钟即可高效获得目标烯烃产物。控制实验显示,镍催化剂和锌粉对于该反应均不可或缺;当缺少其中任一组分时,目标产物均无法形成。此外,该反应不仅适用于芳基碘化物,对于芳基溴化物同样表现出优异反应活性,显示出机械化学条件在促进惰性芳基卤化物转化方面的独特优势。
图2. 条件优化。图片来源:Chin. Chem. Lett.
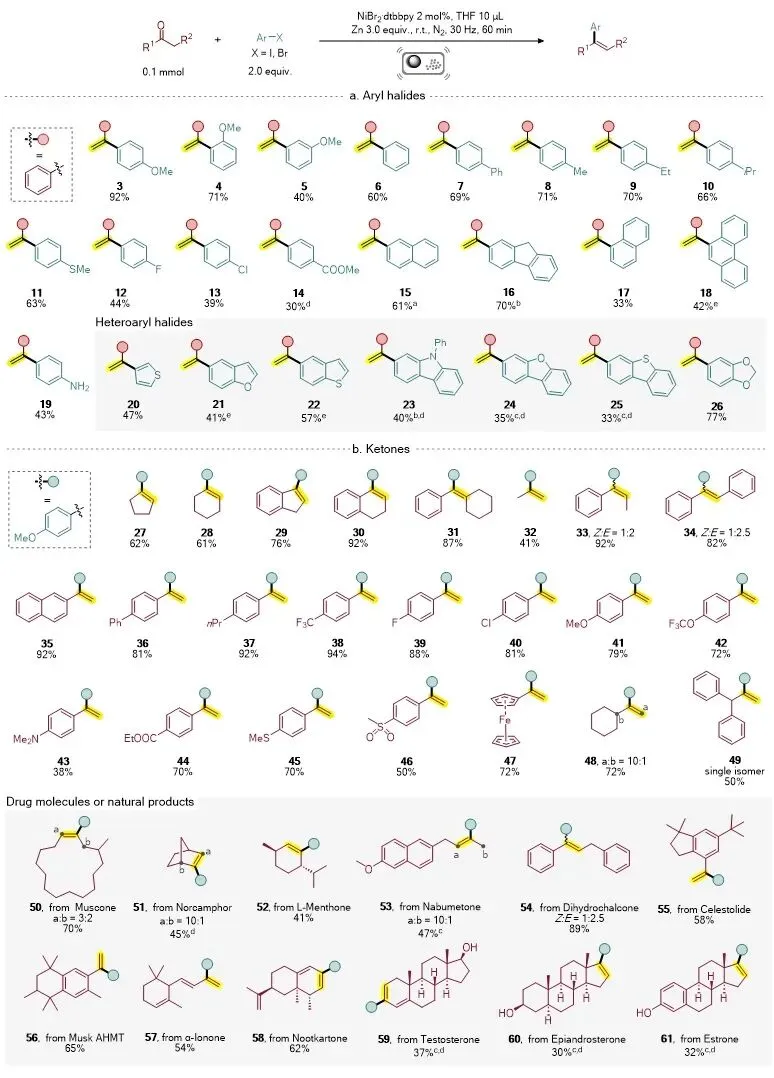
在底物拓展方面,该反应表现出良好的普适性和官能团兼容性。对于芳基卤化物部分,多种芳基碘化物和芳基溴化物均可顺利参与反应,含有烷基、芳基、卤素、酯基、氨基以及共轭芳环等取代基的底物均能转化为相应芳基烯烃产物。此外,部分含氮、含硫和含氧杂芳基卤化物也能够适用于该体系,进一步拓展了该方法在杂环芳基烯烃合成中的应用范围(图3a)。对于酮类底物,研究人员考察了环状酮、非环状酮以及多种官能团化芳基酮。结果表明,环己酮、环戊酮等环状酮具有良好兼容性;非环状酮中,无论芳环上带有给电子基还是吸电子基,均可实现目标转化。含氟、氯、甲氧基、三氟甲氧基、二烷基氨基、酯基、甲硫基、甲砜基和二茂铁等官能团的酮类底物均能有效参与反应,显示出该体系良好的官能团耐受性和后续衍生化潜力(图3b)。
图3. 底物拓展。图片来源:Chin. Chem. Lett.
为了进一步展示该方法的合成实用性,研究人员将该策略应用于复杂天然产物和药物分子相关酮类底物的修饰。麝香酮、L-薄荷酮、萘丁美酮、二氢查耳酮、α-紫罗兰酮、诺卡酮、睾酮、表雄酮和雌酮等复杂结构均可在该体系中实现芳基烯烃化修饰。其中,甾体类酮底物通常由于溶解性差、位阻较大而较难进行选择性修饰,而该机械化学体系能够在少溶剂条件下有效克服这类限制,体现了球磨反应在复杂分子转化中的独特优势(图3b)。
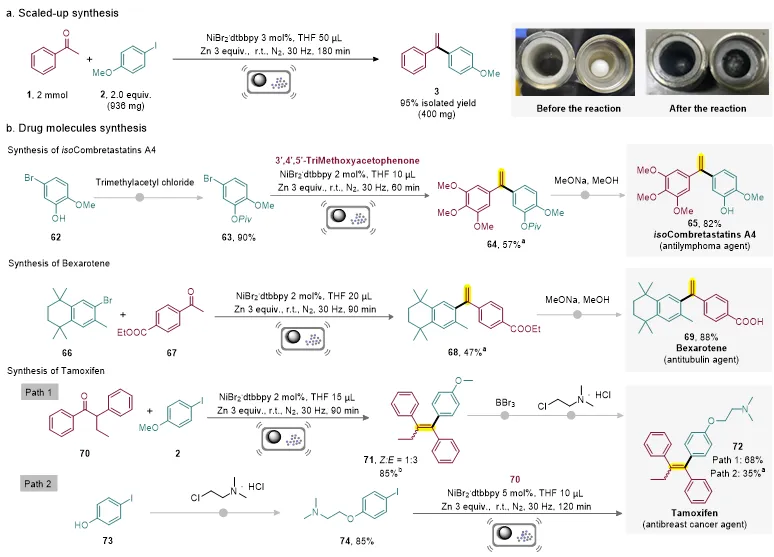
随后,研究人员开展了放大反应和药物分子合成研究。在2 mmol规模下,通过使用ZrO2球磨罐和ZrO2研磨球,模型反应仍能以95%的分离产率获得目标产物,证明了该方法具有较好的放大潜力。进一步地,该策略被用于isoCombretastatin A4、Bexarotene和Tamoxifen等药物分子的合成(图4)。通过该烯烃化方法,可以从简单易得的起始原料出发,高效构建关键烯烃中间体,并经后续转化获得目标药物分子。这些结果表明,该方法不仅适用于模型底物研究,也具有较好的药物分子合成和后期结构修饰价值。
图4. 合成应用。图片来源:Chin. Chem. Lett.
为阐明反应机制,研究人员开展了组分分析、控制实验和密度泛函理论(DFT)理论计算等研究。由于直接研究球磨体系存在一定难度,作者首先比较了球磨体系和溶液体系在相同反应进程下的组分分布(图5a)。GC 分析显示,两种体系中反应物、目标产物和主要副产物具有相似变化趋势,说明二者可能遵循相似的基本反应路径。随后,研究人员制备了芳基镍中间体ArNiIIX,并将其加入反应体系中。结果显示,在锌粉存在下可获得目标烯烃产物,而缺少锌粉时则无法生成产物,说明芳基镍中间体可能是反应中的关键物种,同时锌在反应中并非仅仅作为还原剂存在。
进一步控制实验表明,若直接使用芳基锌试剂与酮反应,并不能检测到醇或烯烃产物,说明该体系中不太可能先经转金属化生成芳基锌试剂,再对羰基进行加成(图 5b)。相反,研究人员更倾向于认为,芳基镍物种直接对被活化的羰基进行亲核加成,形成醇镍中间体,随后与原位生成的ZnX2发生转金属化,形成醇锌中间体,并进一步促进C–O σ键断裂生成烯烃产物。
图5. 机理研究。图片来源:Chin. Chem. Lett.
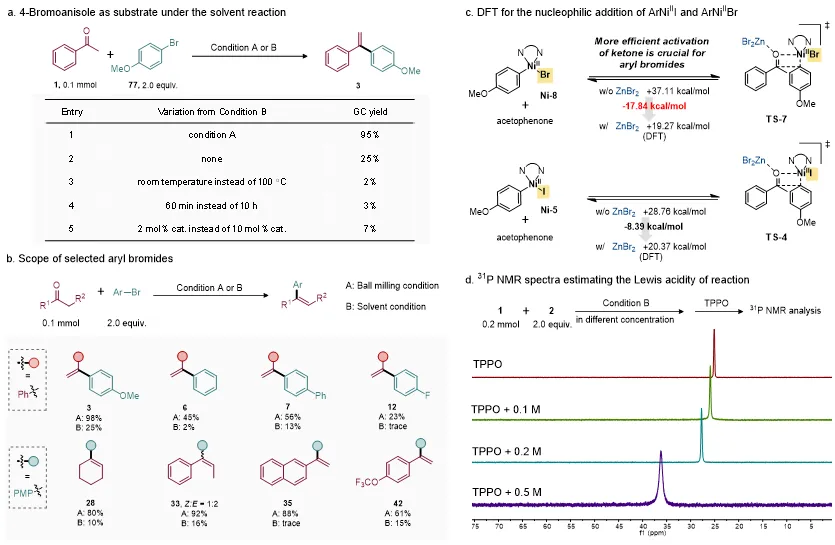
DFT计算进一步支持了上述机制假设。计算结果表明,在ZnBr2作为Lewis酸参与羰基活化时,芳基镍物种对酮羰基的亲核加成过程更为有利;而芳基锌物种直接进攻羰基则具有更高能垒,与实验结果相一致(图5c)。更重要的是,对于芳基溴底物而言,Lewis酸活化羰基对于降低反应能垒尤为关键。相比传统溶液条件,球磨体系具有高浓度或无溶剂的特性,可能有利于减少溶剂分子对ZnX2的竞争配位,从而增强ZnX2对羰基的Lewis酸活化能力(图6c)。
为了进一步验证这一点,研究人员采用Gutmann–Beckett方法评估不同浓度反应体系中的Lewis酸性。结果显示,随着体系浓度升高,三苯基氧膦的³¹P NMR信号逐渐向低场移动,表明Lewis酸络合作用增强(图 6d)。该结果说明,球磨条件下的近无溶剂或高浓度环境能够有效提升羰基活化效率,这也是机械化学体系相较于传统溶液体系表现出更高反应活性的关键原因之一。
图6. 对比实验。图片来源:Chin. Chem. Lett.
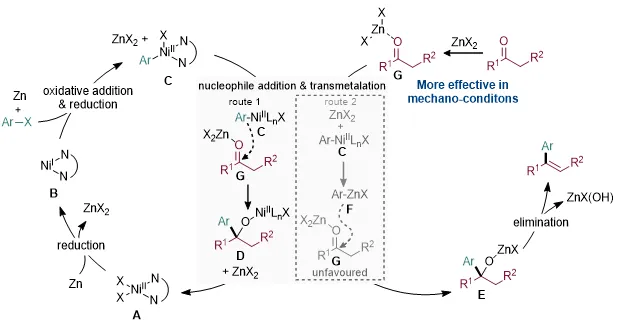
基于上述实验和理论计算,作者提出了合理的催化循环(图7)。首先,NiII物种在锌粉作用下被还原生成低价镍活性物种;随后,芳基卤化物与低价镍发生氧化加成,并经锌还原形成芳基镍中间体,同时生成ZnX2。原位生成的ZnX2作为Lewis酸活化酮羰基,促进芳基镍中间体对羰基的亲核加成,得到醇镍中间体。随后,该中间体与ZnX2发生转金属化,释放NiII物种并形成醇锌中间体,最终经C–O σ键断裂和消除过程生成目标芳基烯烃产物。
图7. 反应催化循环。图片来源:Chin. Chem. Lett.
综上而言,本研究发展了一种机械化学球磨促进的镍催化酮类化合物与芳基卤化物直接还原烯烃化反应。该方法以易得酮和芳基卤化物为原料,在温和、少溶剂条件下实现芳基取代烯烃的高效构建,具有底物范围广、官能团兼容性好、可放大和适用于复杂分子修饰等特点。机制研究揭示了锌在反应体系中的多重作用,即同时作为还原剂、羰基活化剂和C–O σ键断裂促进剂;同时也表明,机械化学条件下增强的羰基活化是实现该高效转化的重要因素。该工作不仅为芳基取代烯烃的绿色合成提供了新方法,也为机械化学促进过渡金属催化还原偶联和惰性化学键转化提供了新的研究思路。
该研究工作近期被Chinese Chemical Letters接收发表。南京师范大学食品与制药工程学院硕士研究生杨兵为文章第一作者,通讯作者为南京大学化学化工学院王毅教授和南京师范大学食品与制药工程学院杜炳南教授。该研究得到了江苏省特聘教授基金(164080H00243)和江苏省合成生物学基础研究中心(BK20233003)的支持。
杜炳南教授课题组长期聚焦机械化学驱动的绿色有机合成研究,致力于以机械力作为非传统能量输入方式,发展区别于传统溶液反应的新型反应模式。课题组重点围绕机械化学条件下的过渡金属催化、惰性化学键活化及高效合成方法学开展系统研究,并进一步探索相关策略在药物分子构建、创新药研发和靶向诊断分子设计中的应用,旨在建立更加绿色、简洁且具有实用价值的有机合成新体系。近五年来,以第一作者或通讯作者在J. Am. Chem. Soc.、Nat. Commun.、ACS Catal.、JACS Au、Green Chem.、Commun. Chem.、Org. Lett.等期刊发表论文十余篇。
原文(扫描或长按二维码,识别后直达原文页面):

Ball milling-promoted nickel-catalyzed reductive olefination of ketones with aryl halides
Bing Yang, Tingrui Liu, Nan Liu, Yi Wang, Bingnan Du
Chin. Chem. Lett., 2026, DOI: 10.1016/j.cclet.2026.112858
点击“阅读原文”,查看 化学 • 材料 领域所有收录期刊