南京大学瞿晓磊/付翯云&厦门大学王野/吴雪娇,最新JACS!精准硒掺杂解锁钯纳米团簇以实现高效化学选择性加
- 2026-05-30 00:14:38


第一作者:胡晓洁
通讯作者:瞿晓磊、王野、付翯云、吴雪娇
通讯单位:南京大学、厦门大学
卤代苯胺是医药、农药、染料工业的核心中间体,传统热化学还原(铁粉法)会产生大量废渣、能耗高、污染严重。多相催化加氢是绿色替代路径,但高活性钯(Pd)催化剂易过度加氢,导致脱卤副反应,降低目标产物选择性;同时 Pd 易因硫、卤素中毒失活,稳定性差。亚纳米 Pd 团簇原子利用率高、活性强,但与载体强相互作用易使 Pd 呈高价态(Pd^δ+),选择性与抗中毒性进一步下降。
针对 “活性 - 选择性 - 稳定性” 三重矛盾,本研究提出精准硒(Se)掺杂新策略:通过液相催化加氢法,在缺陷二氧化钛(dTiO₂)负载的全暴露 Pd 团簇表面定点沉积微量 Se,调控 Pd 电子态,平衡加氢活性与选择性,同时增强抗硫中毒能力,为卤代芳烃绿色选择性加氢提供高效、稳定、低成本新方案。
1. 位点特异性硒掺杂策略
该研究首次提出了一种通过液相催化氢化法,在室温下将硒(Se)精确、位点特异性地沉积于完全暴露的钯(Pd)纳米团簇表面。与传统的浸渍法导致Se随机掺杂不同,该方法实现了Se仅在Pd活性位点上精准修饰,有效避免了Pd位点被过度堵塞,并显著提高了Pd-Se配位数,从而克服了传统掺杂方法中掺杂位置不可控的难题,实现了对金属纳米团簇表面结构的原子级精准调控。
2. 电子富集效应破解活性-选择性矛盾
研究发现,痕量Se的精准掺杂能够向Pd位点有效转移电子,形成电子富集的Pd⁰态。这一电子结构的调控产生了双重关键作用:一方面增强了Pd对H₂的活化能力,保持了高催化活性;另一方面显著抑制了底物中卤素原子的非特异性吸附,从而完全抑制了副反应(氢脱卤反应)。最终,该催化剂在多种卤代硝基苯的加氢反应中,实现了>99%的卤代苯胺选择性、15,593 h⁻¹的高转化频率,以及优异的抗毒化与循环稳定性,成功打破了传统负载型金属催化剂中高活性与高选择性难以兼得的瓶颈。
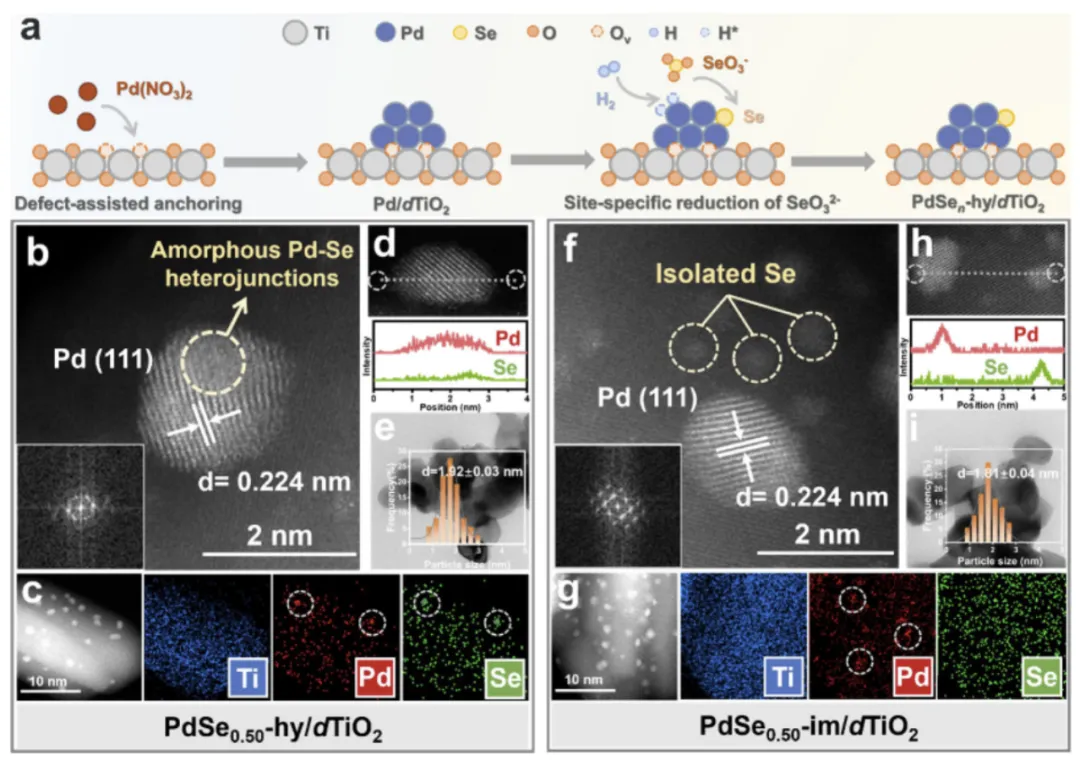
图 1 催化剂合成与微观结构表征
图 1a 为合成策略:dTiO₂负载全暴露 Pd 团簇(平均 0.93 nm、分散率 97.4%),再通过液相加氢沉积 Se,形成 PdSeₙ-hy/dTiO₂;对照样为传统浸渍法制备 PdSe₀.₅₀-im/dTiO₂。图 1b-c AC-HAADF-STEM 与 EDS 映射显示,PdSe₀.₅₀-hy/dTiO₂中 Se精准包覆 Pd 表面、均匀分布;图 1d 线扫描证实 Se 与 Pd 共定位;图 1e 粒径随 Se 掺杂量递增(0.93→2.38 nm);图 1f-h 显示传统浸渍法 Se 随机分布、部分脱离 Pd,证明定点沉积法精准可控。
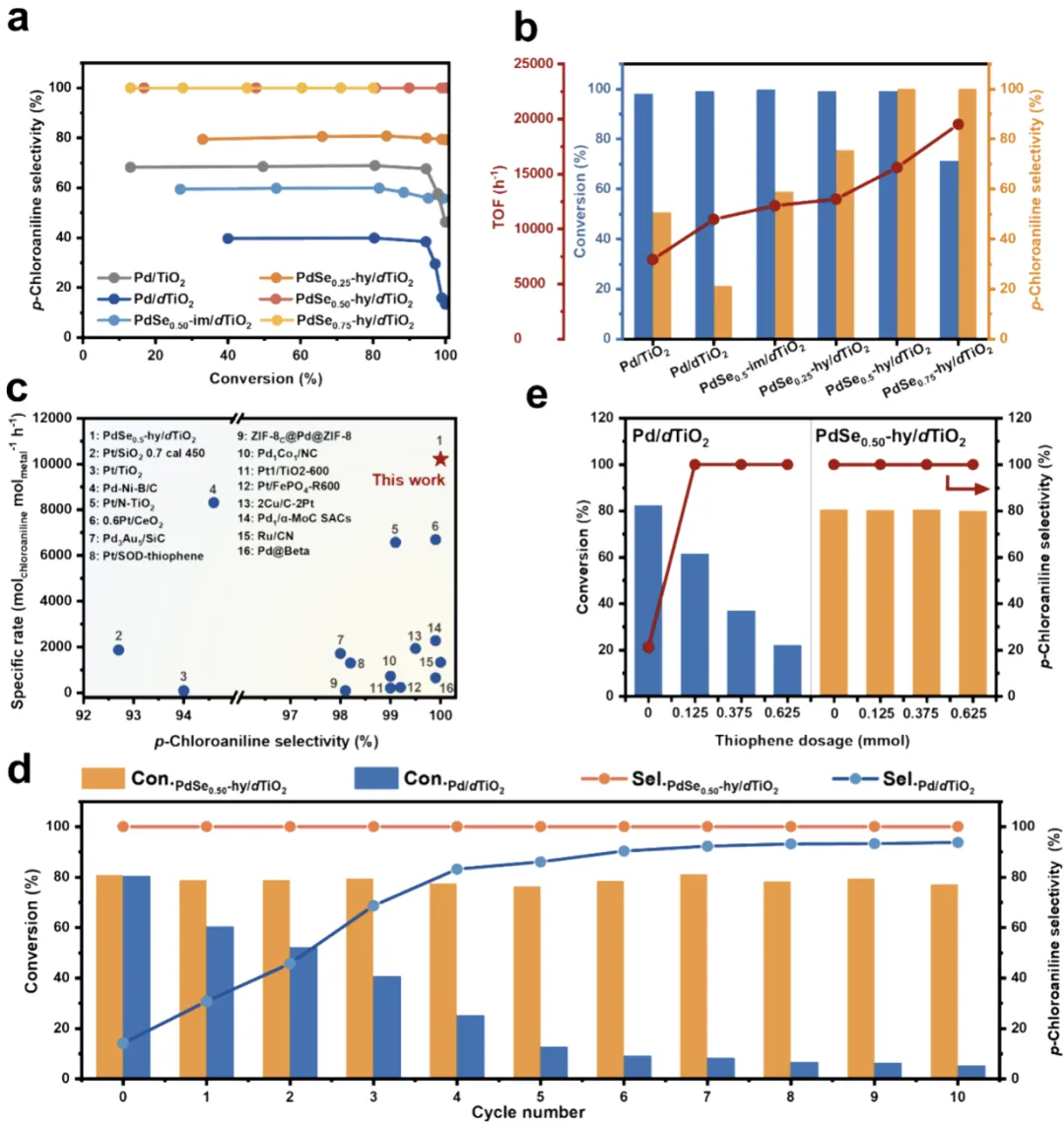
图 2 催化性能评价
图 2a 选择性 - 转化率曲线:纯 Pd/dTiO₂选择性仅 15.8%,Se 掺杂后选择性骤升,PdSe₀.₅₀-hy/dTiO₂完全转化下选择性 > 99%;图 2b 活性对比:PdSe₀.₅₀-hy/dTiO₂的 TOF(15,593 h⁻¹)显著高于纯 Pd 与传统掺杂样;图 2c 性能对比:远超文献贵金属催化剂;图 d 循环:纯 Pd 10 循环后活性骤降,PdSe₀.₅₀-hy/dTiO₂10 循环后活性 / 选择性几乎不变;图 e 抗硫性:高浓度噻吩下性能稳定,证实富电子 Pd 抗中毒优势。
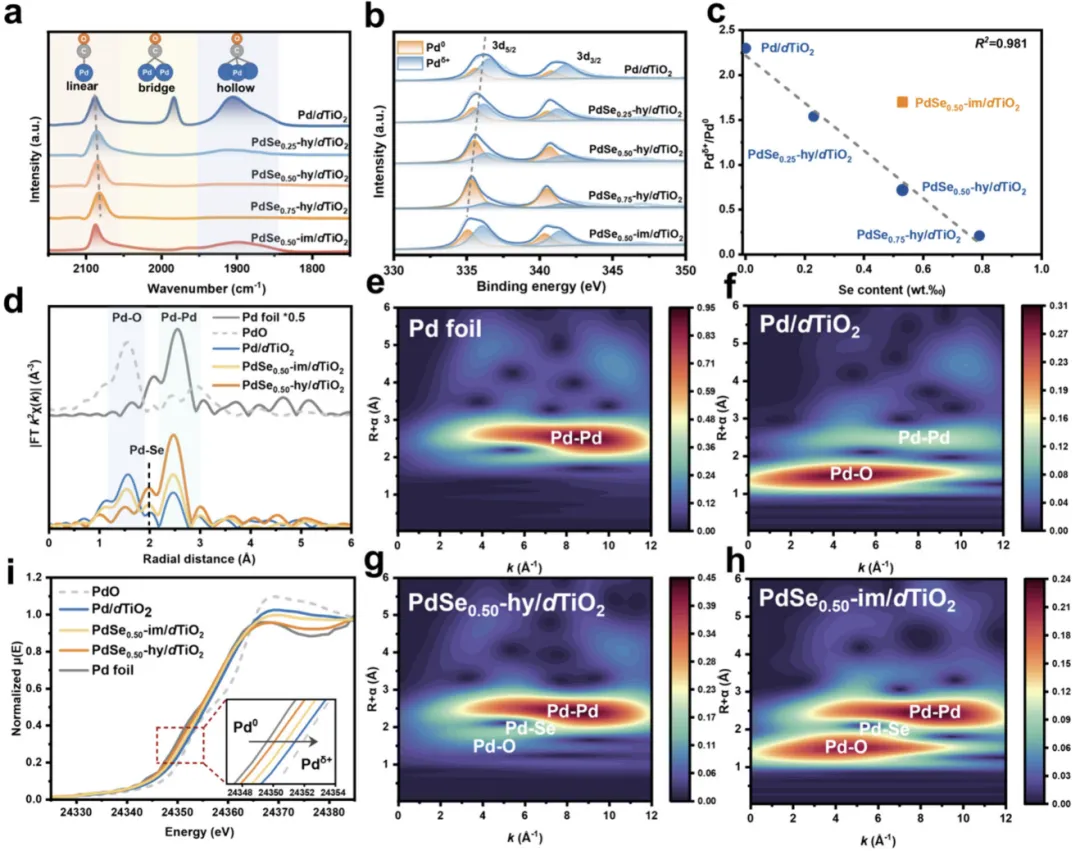
图 3 电子态与界面结构解析
图 3a 原位 CO-DRIFTS:纯 Pd 以桥 / 空心吸附为主,Se 掺杂后转为线性吸附,表明连续 Pd 位点被 Se 隔断;图 3b XPS:Se 掺杂使 Pd 3d 峰向低能偏移,Pd⁰比例升高、Pd^δ+ 降低;图 3c Pd^δ+/Pd⁰比随 Se 掺杂量线性下降;图 3d-h EXAFS:PdSe₀.₅₀-hy/dTiO₂存在Pd-Se 键、无 Pd-Pd 团聚,Se 配位更高,证实强电子转移、精准界面结构。
本研究开发液相定点 Se 掺杂策略,在 dTiO₂负载全暴露 Pd 团簇表面精准沉积微量 Se,调控 Pd 电子态为富电子态(Pd⁰为主),平衡加氢活性与选择性。PdSe₀.₅₀-hy/dTiO₂对卤代硝基苯加氢选择性 > 99%、TOF 达 15,593 h⁻¹,完全抑制脱卤副反应;同时具备优异抗硫中毒性与循环稳定性。
机理研究证实:Se 供电子抵消载体强相互作用,弱化卤代苯胺吸附、强化脱卤能垒,实现选择性调控;富电子 Pd 抑制硫 / 卤素吸附,增强稳定性。该工作突破传统催化剂 “活性 - 选择性 - 稳定性” 矛盾,为精细化工绿色选择性加氢提供精准电子调控新范式,具有重要工业应用价值。
文献链接:
https://doi.org/10.1021/jacs.6c04939
以上内容,如有误读和纰漏,敬请指正
解决世纪争议难题,郑州大学“最硬核团队”重磅Nature: 合成纯相块状六方型金刚石 院士携手!兰州大学,校史首篇Nature Nanotechnology:飞秒时间尺度上的质子 - 电子时间异步性,实现质子交换膜电解槽用耐腐蚀低铱阳极 分子筛催化,再发Nature Catalysis! 南开大学陈军院士,2025年成果汇总 最新JACS,高稳定负载型 Cu-Ni 稀释合金催化剂实现高效 CO2 重整甲烷 中科大,最新Nature Chemistry!双原子催化剂! 杰青联手!华东师范大学关小红/孙远奎&湖南大学王双印,最新Angew:硫掺杂零价铁气凝胶*NO偶联和*H供给协同调控驱动硝酸盐高效电还原成N2 湖南大学「国家杰青」王双印&中南大学任博华&天津大学杨娜,最新Angew:机器学习揭示有机-金属界面“电子海绵”行为促进CO2电还原C2+生成 李灿院士领衔!兰州大学李泽龙,最新Angew:NdO强化IrMnOx双位点协同,酸性OER低Ir长寿命! 韩布兴院士领衔!中科院化学所张裴,最新Angew!温度介导动力学与界面控制以实现浓缩热敏性生物质原料持续电氧化!

如需转载或合作,请联系我们
联系方式:15715750735(微信同)
联系邮箱:mon@xueyanhui.com
2.催化进展现有综合群、电催化交流群、同步辐射交流群、文献交流互助群、各研究领域群等近20余个,欢迎大家加小编微信,我们会尽快拉您进入对应的群。

随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- 南京89年公务员小姐姐|本科164|有房有车|期待173-183、30-35岁、真诚耐心的你
- 南京鼓楼丨省20万买67平精装两房 双南地铁
- 500个大混战 全南京对比 最后他居然选了这里
- 南京政区地名历史文化解读之(4)南京最突出的名号是为金陵
- 南京北站转地上施工,16台30线69.21万平米计划2027年通高铁
- 暴雨!6级阵风!南京气象刚刚发布
- 欢迎南京的帅哥美女们来新街口梦健舞艺术学校学习摩登舞
- 水游城惊现“清代双胞胎水井”,竟是老南京的“共享饮水机”!
- 深化校地产学研融合:南京农业大学苏州校友会走进姑苏实验室开展对接交流
- 【南京长江医院心理科】焦虑情绪与焦虑症:如何区分正常与异常?
