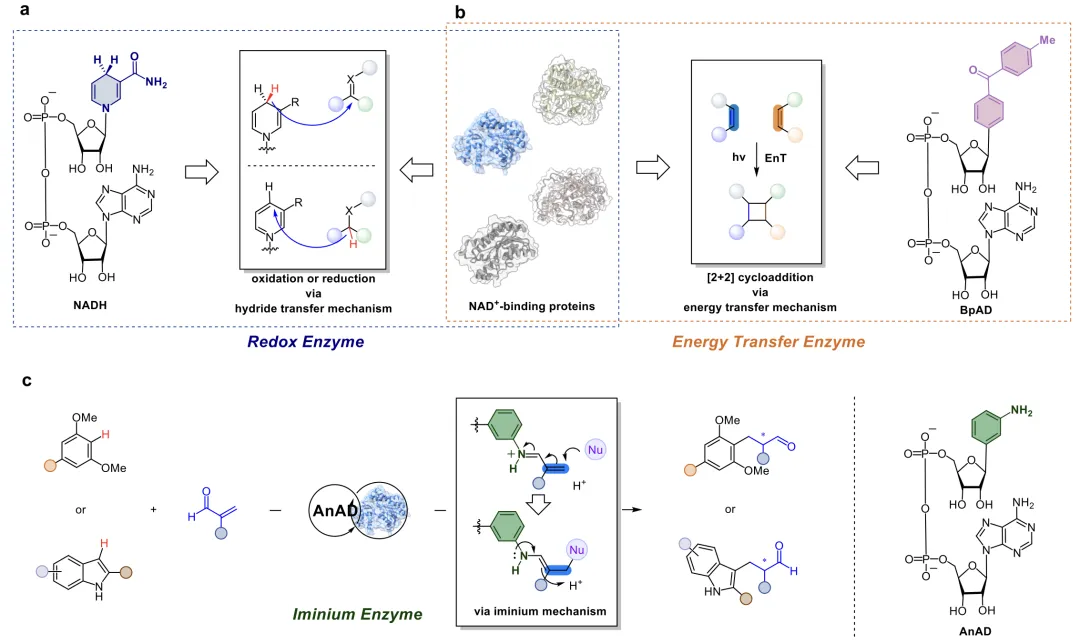
人工酶旨在融合生物催化的高选择性、可进化性与化学催化多样的反应活性,而将非天然催化基团整合到蛋白骨架中是领域核心难题。目前主流有两种技术路线:一是嵌入功能化非天然氨基酸,但该方法需配套专属正交翻译系统,还会导致蛋白表达量下降,不利于规模化;二是引入人工辅因子,依靠共价连接、金属配位或超分子作用结合蛋白,却普遍存在结合方式有限、特异性弱、适配骨架少等缺陷。因此,开发操作简单、普适性强、便于定向进化优化的辅因子体系,是提升人工酶平台实用性的关键。
近日,南京大学潘惠杰课题组在Angewandte Chemie International Edition上发表研究,开发出一款全新NAD⁺型人工辅因子,成功改造广泛存在的NAD(P)⁺结合蛋白,使其具备全新的亚胺离子催化能力,该策略核心思路是基于该本课题组前期开发了一种光活性辅因子BpAD(二苯甲酮腺嘌呤二核苷酸),该NAD+类似物通过将烟酰胺基团替换为光敏剂实现功能改造。BpAD的引入可有效地将多种NAD+结合蛋白重塑为人工光酶,这些人工光酶可通过能量转移机制实现[2 + 2]环加成反应(Nat. Catal., 2025, 8, 822)。随后通过对BpAD进行修饰改造,开发了新型光活性辅因子MeO-BpAD,成功实现了对映选择性的分子间[2π + 2σ]环加成反应(J. Am. Chem. Soc. 2026, 148, 13755)。基于这一研究思路,研究团队现拓展烟酰胺腺嘌呤二核苷酸型辅因子的催化应用范围,通过将烟酰胺部位替换成具有催化活性的苯胺,开发了一种新的辅因子AnAD,成功将天然的氧化还原酶重编程为亚胺离子人工酶,在生物催化领域首次实现了非天然的串联Friedel-Crafts烷基化-对映选择性质子化反应(图1)。
图1 基于NAD⁺构建的人工酶
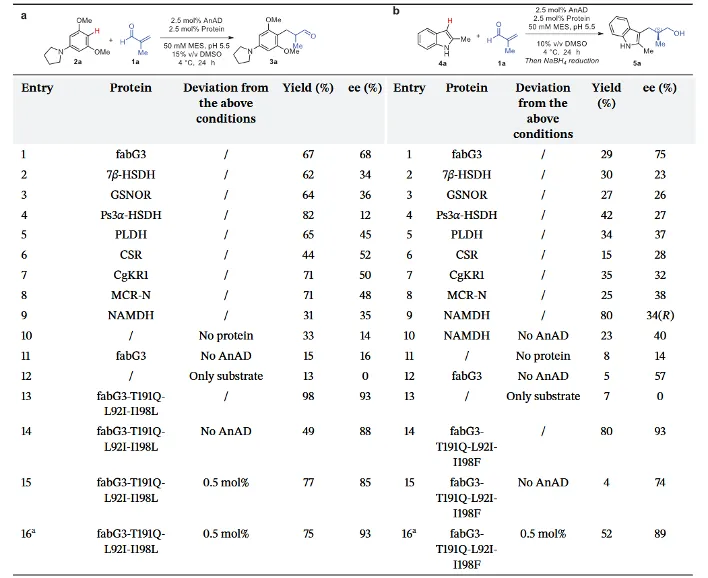
研究团队使用富电子芳烃2a与甲基丙烯醛1a作为模板反应,测试了由多种野生型NAD(P)+依赖性蛋白组成的支架库,实验结果表明,不同来源的蛋白支架兼具宽泛的本征兼容性,同时在催化活性与立体选择性上呈现出显著差异。其中,结核分枝杆菌来源的脱氢酶变体fabG3展现出最优的初始催化性能(67%产率,68% ee)。后续对照实验进一步验证,单一的辅助因子AnAD或野生型蛋白支架均无法实现高效的立体控制,只有二者协同组装形成的全酶体系后,才能高效诱导高立体选择性的催化转化。这一结果充分证实,人工辅助因子与蛋白微环境的协同作用,对体系催化活性与手性诱导效果起到了决定性调控作用。
表1 反应条件优化与蛋白改造
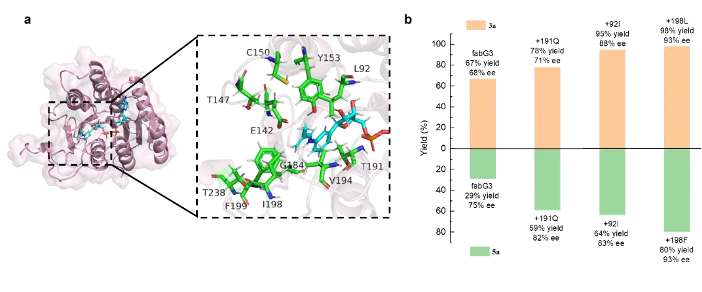
为了进一步优化该催化体系,研究团队通过分子对接技术将1a和AnAD形成的亚胺中间体对接至fabG3活性位点中(PDB: 1NFQ)。结构模型显示,AnAD的结合模式与天然辅酶高度一致,其苯胺单元精准地指伸入了底物结合口袋(如图2a所示)。基于该结构模型的指导,研究团队对距离中间体5Å以内的关键氨基酸位点进行定向进化。通过三轮进化改造,最终成功筛选出了两个最优突变体(fabG3-T191Q-L92I-1198L与fabG3-T191Q-L92I-1198F),将两类反应底物的化学产率和对映选择性分别最高提升至98%和93% ee以及80%和93% ee(如表1所示)。值得一提的是,该进化改造后的蛋白在未添加AnAD辅因子时,依旧表现出良好的催化性能,产物收率49%,对映体过量值达88%。这说明氨基酸突变重塑了活性口袋的空间结构,有效优化了底物的结合方式与空间取向。此外,该全酶体系展现出了优异的有机溶剂耐受性,在高达30% v/v的DMSO共溶剂存在下依然能保持极高的催化活性。
图2 蛋白的结构建模与定向进化
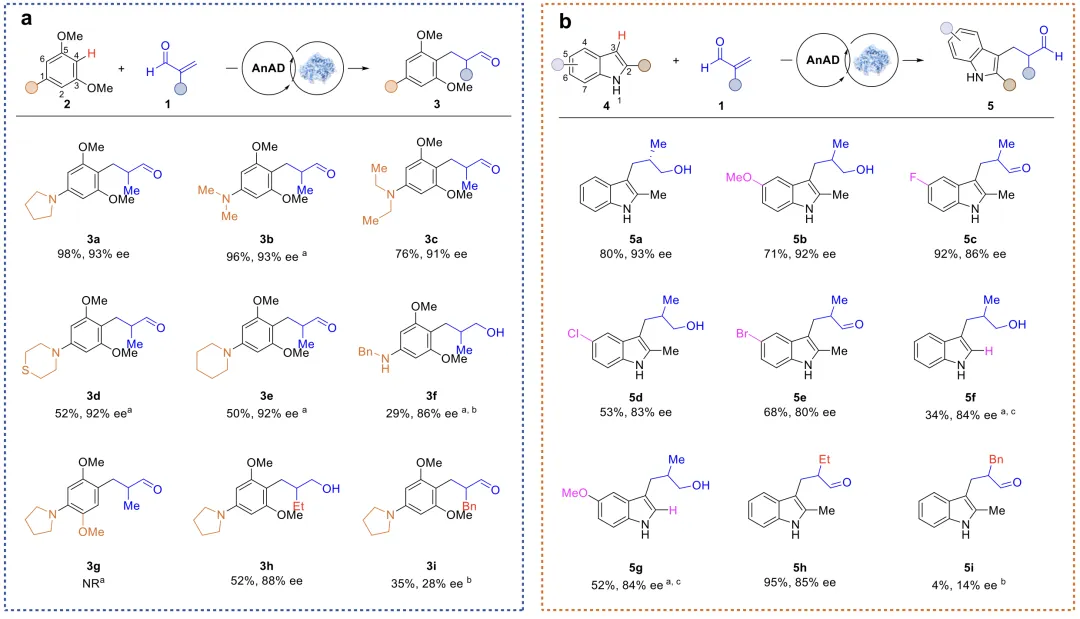
在最优反应条件下,对底物适用性进行考察,小体积二烷基氨基取代底物的反应收率普遍较高(3b-3c);当取代基由二甲氨基依次替换为哌啶基、空间位阻持续增加时,反应收率呈梯度下降,但对映选择性几乎不受影响(3b–3e)。苄胺基取代则会导致收率大幅降低,主要归因于其给电子能力较弱,致使芳烃C4位亲核活性显著下降。而烯醛底物的空间位阻耐受性有限,将α位取代基由甲基替换为乙基(3h)后,反应活性下降,对映选择性也略有降低;而引入体积更大的苄基(3i)时,产物收率与立体选择性均大幅下降。若将烯醛底物1替换为结构相近的甲基乙烯基酮,则完全没有反应活性,这大概率是因为酮类底物形成相应亚胺离子中间体的能力有所下降。对于吲哚类底物,无论其芳环带有给电子基团还是吸电子基团,反应均可顺利进行(5b–5e)。研究表明,2-甲基基团发挥着关键作用,主要归因于其可显著增强吲哚C3位的亲核活性;若移除该基团,反应转化率将大幅下降(5f),而在芳环上引入额外给电子基团,可在一定程度上补偿这一活性损失(5g)。同时,烯醛底物1的α位空间位阻显著影响反应活性与对映选择性:乙基取代底物可正常参与反应,而大位阻苄基取代底物则完全无法转化。此外,甲基乙烯基酮在该体系中同样无反应活性。
图3 底物的普适性研究
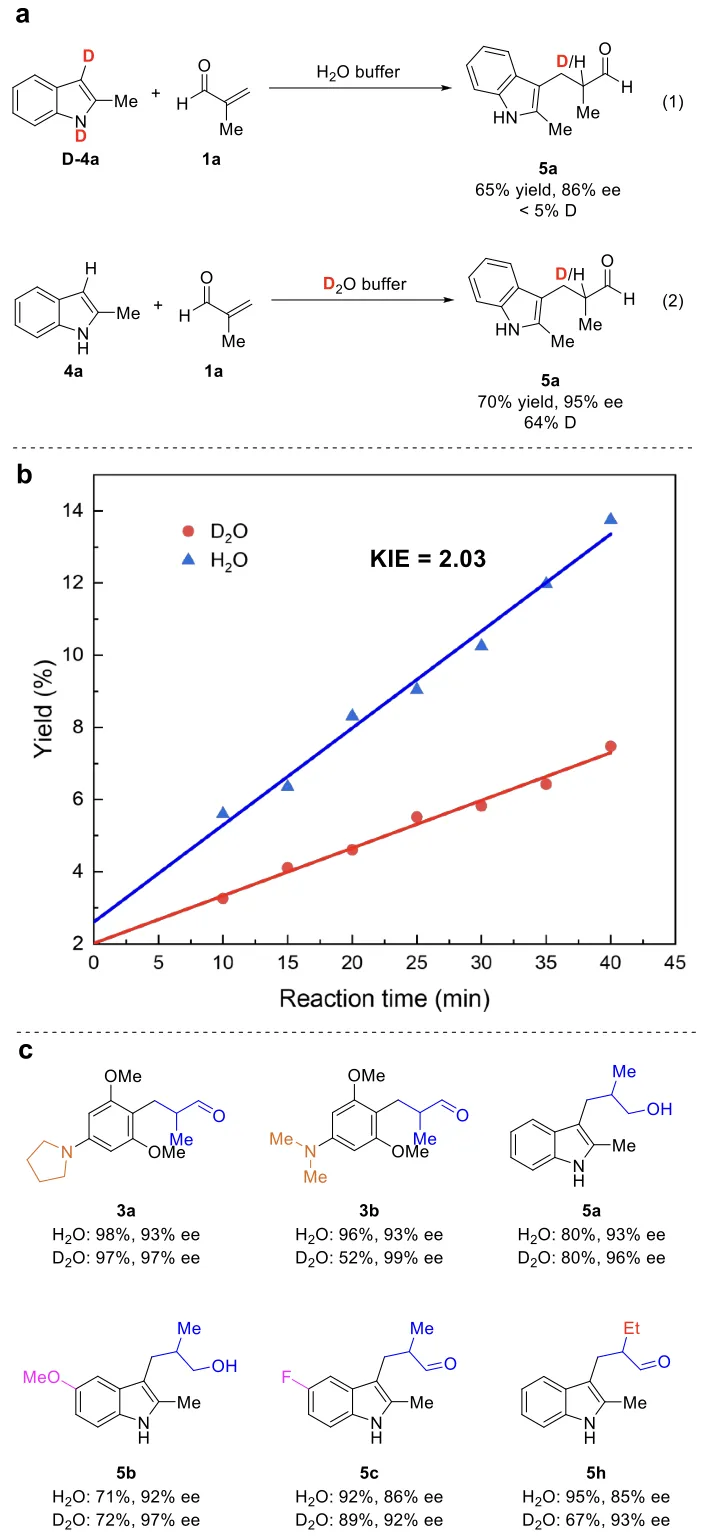
为揭示该体系高立体选择性的反应机制,研究团队开展了同位素标记与动力学同位素效应实验。首先进行了同位素标记实验(图4):在标准反应条件下,以氘代吲哚D-4a为底物时,产物氘掺入率低于5%(图4a,反应式1);而以非氘代底物4a在重水体系中反应时,产物氘掺入率可达64%(图4a,反应式2)。该结果明确证实,反应的α位质子来源于反应体系中的水相溶剂,而非底物分子本身。同时,实验测得底物1a与2a、1a与4a反应的动力学同位素效应(KIE)值分别为2.03和1.33(图4b、图S4及表S14),说明质子转移在上述反应的动力学过程中起到关键作用。
图4 机理研究
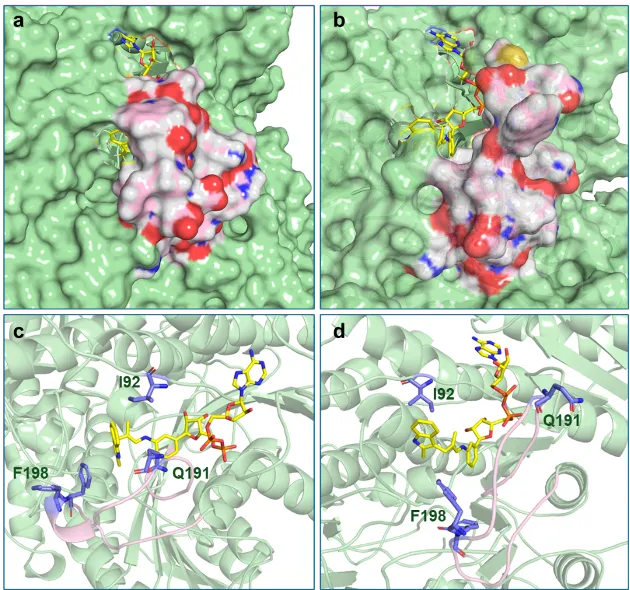
随后,研究人员针对进化后的突变体蛋白开展了分子动力学(MD)模拟。模拟结果表明,该人工全酶在催化过程中存在“互变”的两种主要构象状态。闭合构象下,AnAD的腺苷二磷酸核糖主链占据经典NAD⁺结合口袋,烯胺单元嵌于催化口袋并紧邻T191Q、L92I、F198残基。该构象形成密闭疏水空腔,既阻碍水分子进入以抑制质子化,又能预组织底物1a与4a至优势构型促进傅克烷基化;空腔的大位阻也解释了烯醛底物对大取代基的低兼容性(图5、图S6)。开放构象中,AnAD主链仍固定于NAD⁺结合区,柔性环外摆带动烯胺小幅旋转,使其Re面暴露于溶剂。水分子由此对Re面发生选择性质子化,生成观测到的S构型产物。模拟结果证实了动态催化机制:柔性环的构象变化同时调控催化效率与立体选择性,其中开放构象主导对映选择性质子化过程。
该研究团队开发了一种NAD⁺型辅因子AnAD,可将多种NAD(P)⁺结合蛋白重编程为亚胺离子生物催化剂,实现串联傅克烷基化-对映选择性质子化反应。通过系统筛选蛋白骨架,获得了多个立体化学结果可调的功能骨架;后续蛋白工程改造显著提升了催化效率与对映选择性,并拓展了底物范围,可兼容多种富电子芳烃及吲哚类衍生物。机理研究表明,反应活性与立体控制源于AnAD与蛋白微环境的协同作用,其中柔性环的动态运动主导了对映选择性质子化步骤。该工作进一步证实,合成NAD⁺型辅因子是一类稳健且通用的工具,可拓展广泛存在的NAD(P)⁺结合蛋白的催化功能,从而实现新的非天然生物转化。

 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
