南京大学「国家优青」金钟最新AM | 调控α-MoC₁₋ₓ负载钌团簇与单原子实现高效水分解!
- 2026-06-15 13:18:17

在保持高效催化活性的同时,最大限度地提高活性金属的利用率,对于电催化碱性析氢反应至关重要。

在本文中,作者报道了一种简便的热解策略,将钌簇及相邻的钌单原子锚定在α-碳化钼涂层碳纳米球上(简称为RuCS/SA/α-MoC1-x/C)。密度泛函理论(DFT)计算与原位表征共同揭示了一种电子桥接机制:Ru单原子向缺陷α-碳化钼提供电子,随后电子转移至钌簇,实现了对不同类型钌位点电子结构的协同调控。因此,钌单原子与α-碳化钼的双重激发削弱了钌簇与氢中间体的结合强度,加速了氢气的脱附过程。所制备的3%-RuCS/SA/α-MoC1-x/C样品在10 mA cm-2电流密度下表现出9 mV的优异过电势,其质量活性达20.38 A mg-1Ru(-100 mV条件下),且在25 mV过电势下的转换频率为1.71 H2 s-1,性能优于20% Pt/C催化剂。此外,采用该材料作为阴极电催化剂的阴离子交换膜水电解槽与锌-水电池均展现出卓越的性能。
氢能作为最具潜力替代化石燃料的绿色能源之一,对减少空气污染和缓解气候变化具有重要意义。目前,电催化析氢反应(HER)被认为是最清洁的制氢技术,可利用太阳能、风能和潮汐能等可再生能源产生的绿色电力可持续地产氢。碱性HER作为经济可持续地生产高纯氢的关键工业方法,已成为广泛科学研究的焦点。然而,碱性HER的产氢效率低于酸性HER,主要是由于额外的Volmer步骤(H2O + e- → Had + OH-)被认为是碱性HER的速率决定步骤。
尽管在用于碱性HER的贵金属铂基催化剂方面取得了显著进展,但其高成本仍然是实际应用的关键障碍。因此,开发高效且廉价的碱性HER催化剂至关重要。钌(Ru)的成本仅为铂的约30%,同时表现出与Pt相当的水结合能,成为HER中Pt的理想替代品。迄今为止,尽管Ru基电催化剂在酸性HER中已取得显著进展,但其在碱性HER中的催化活性仍因缓慢的水解离而受到严重制约,从而导致性能不佳。根本挑战在于质子可用性不足,这源于Volmer步骤中H2O解离缓慢以及生成的OH*吸附过强。这些因素协同作用导致催化剂中毒。因此,合理设计高效的Ru基碱性HER电催化剂以媲美酸性HER性能是一项关键挑战。
最近,He等人揭示了嵌入镍空位的Ru单原子导致Ru位点的局域结构极化,降低了水的解离能并改善了氢吸附自由能,该组合在碱性HER中获得了54 mV@10 mA cm-2的性能。此外,Hu等人发现Ru团簇的电子结构具有尺寸依赖性:与Ru单原子相比,1 nm Ru团簇表现出优异的H2O解离能力和上移的d带中心,而3 nm Ru团簇则因优化的氢吸附-脱附动力学而表现出增强的HER活性。
Zhang等人进一步提出,在LaRuSi3表面上电化学触发Ru团簇再生可诱导优化的电荷离域,从而促进Ru活性位点的水吸附-解离,同时增强电导率和电化学表面积。这些发现共同揭示了钌基材料在碱性HER中独特的尺寸效应和界面调控机制。基于此本文推测Ru团簇与单原子结合可能更有利于水分子解离。然而,OH*中毒以及单原子易团聚成更大团簇等问题仍然是巨大挑战。
大量研究表明,合适的载体不仅能锚定金属原子/团簇以抑制团聚,还能通过强金属-载体相互作用(SMSI)优化负载金属的电子结构,从而调控OH*吸附能垒以缓解中毒效应。金属碳化物具有优异的稳定性、出色的导电性和耐腐蚀性。此外,碳的电负性低于O、S、Se和P元素,这有助于优化Ru的电子结构,使更多d电子参与HER。值得注意的是,具有丰富空位的α-MoC1-x表现出本征HER活性并展现出显著潜力。此外,理论分析表明α-MoC1-x可显著加速Volmer步骤动力学,从而提高HER速率。然而,受限于α-MoC1-x的合成挑战,利用其作为载体锚定Ru单原子和团簇的探索性研究极为罕见,这直接导致它们之间的协同机制尚未阐明。
在本文中,作者报道了一种简便的热解策略,使Ru团簇和相邻单原子锚定在α-MoC1-x载体上。受益于碳骨架的空间限域和保护效应,尺寸约5 nm的α-MoC1-x纳米晶被紧密包覆在碳基质中。富含空位的α-MoC1-x作为独特载体,不仅稳定了Ru单原子和团簇(标记为RuCS/SA/α-MoC1-x/C),还通过其丰富的缺陷位点直接参与电荷再分配。高角环形暗场扫描透射电子显微镜(HAADF-STEM)结果证实Ru团簇和单原子成功锚定在α-MoC1-x上。密度泛函理论(DFT)计算首次预测了电子桥接机制:Ru单原子向缺陷α-MoC1-x供电子,后者随后将电子转移至Ru团簇,从而实现对不同Ru位点电子结构的协同调控。该互补机制进一步得到XPS和XAFS分析的证实,验证了单原子与团簇之间相反的电荷转移方向与DFT预测一致。
此外,通过系统改变钌负载量,本文确定了3%是单原子与团簇平衡共存的最佳组成比例。所制备的3%-RuCS/SA/α-MoC1-x/C在碱性溶液中表现出9 mV@10 mA cm-2的优异过电位,以及20.38 A mg⁻¹Ru(-100 mV)的质量活性和25 mV下1.71 s-1的转换频率,这些数值均显著优于20% Pt/C催化剂。同时,使用3%-RuCS/SA/α-MoC1-x/C作为阴极催化剂的阴离子交换膜水电解(AEMWE)池可在1.0 A cm-2电流密度下维持550 h。此外,3%-RuCS/SA/α-MoC1-x/C被用于构建Zn-H2O电池,在1 m KOH中实现持续发电。所组装的电池达到22.50 mW cm-2的功率密度,并能在10 mA cm-2下稳定放电20 h,这归因于3%-RuCS/SA/α-MoC1-x/C催化剂优异的碱性HER性能。本工作不仅通过反向捕获策略在原子水平上有效提升了催化剂的催化活性,也为构建高效的AEMWE池和Zn–H2O电池开辟了更多机会。
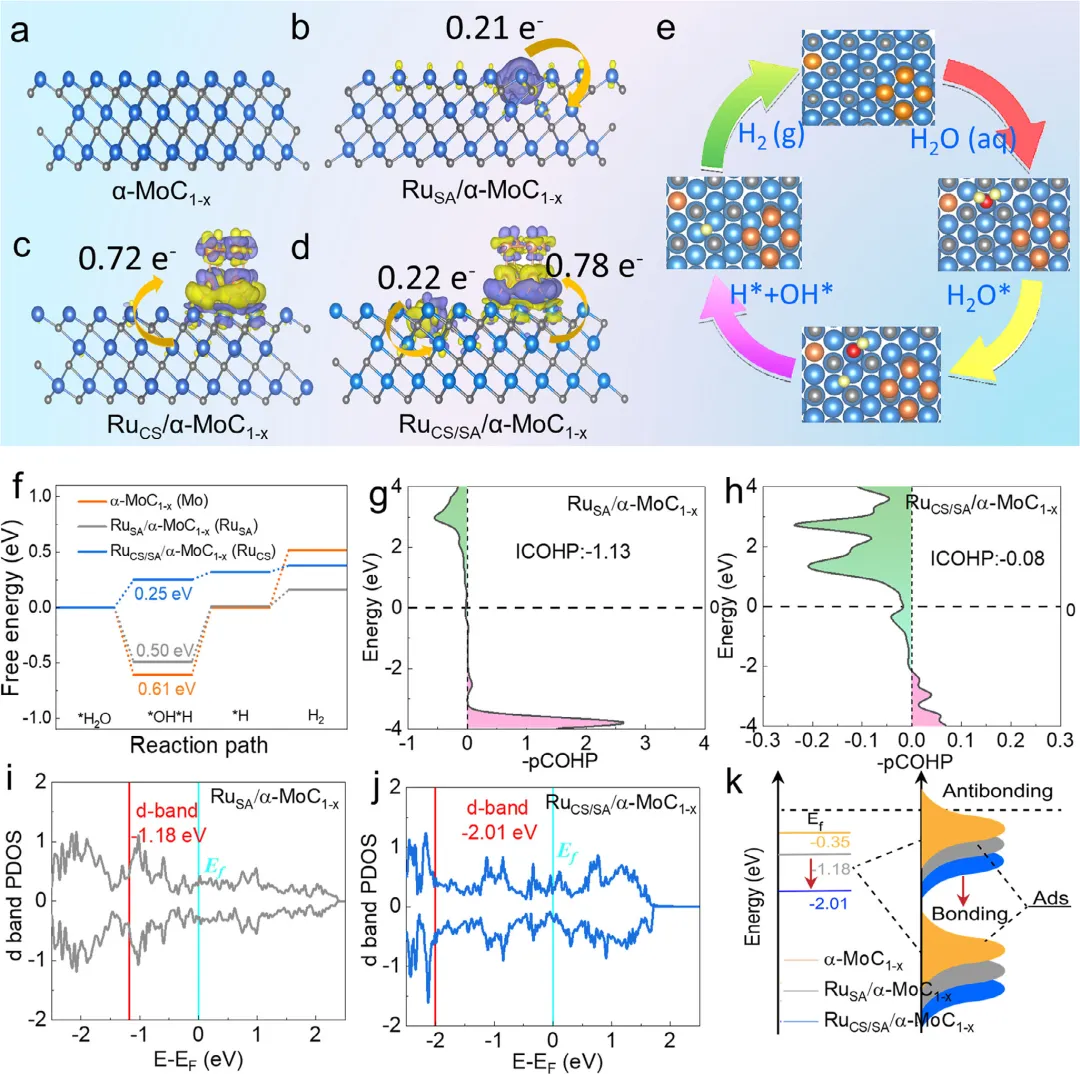
图1:a–d) α-MoC1-x、RuSA/α-MoC1-x、RuCS/α-MoC1-x和RuCS/SA/α-MoC1-x的差分电荷密度(等值面单位0.007 eÅ,浅蓝色、灰色和橙色球分别代表Mo、C和Ru)。e) 主要反应步骤的中间产物。f) α-MoC1-x、RuSA/α-MoC1-x和RuCS/SA/α-MoC1-x/C模型上完整HER反应的反应能量分布图。g和h) RuSA/αMoC1-x和RuCS/SA/α-MoC1-x的ICOHP值。i,j) RuSA/α-MoC1-x和RuCS/SA/α-MoC1-x/C的d带PDOS。k) α-MoC1-x、RuSA/α-MoC1-x和RuCS/SA/α-MoC1-x/C的d带中心。
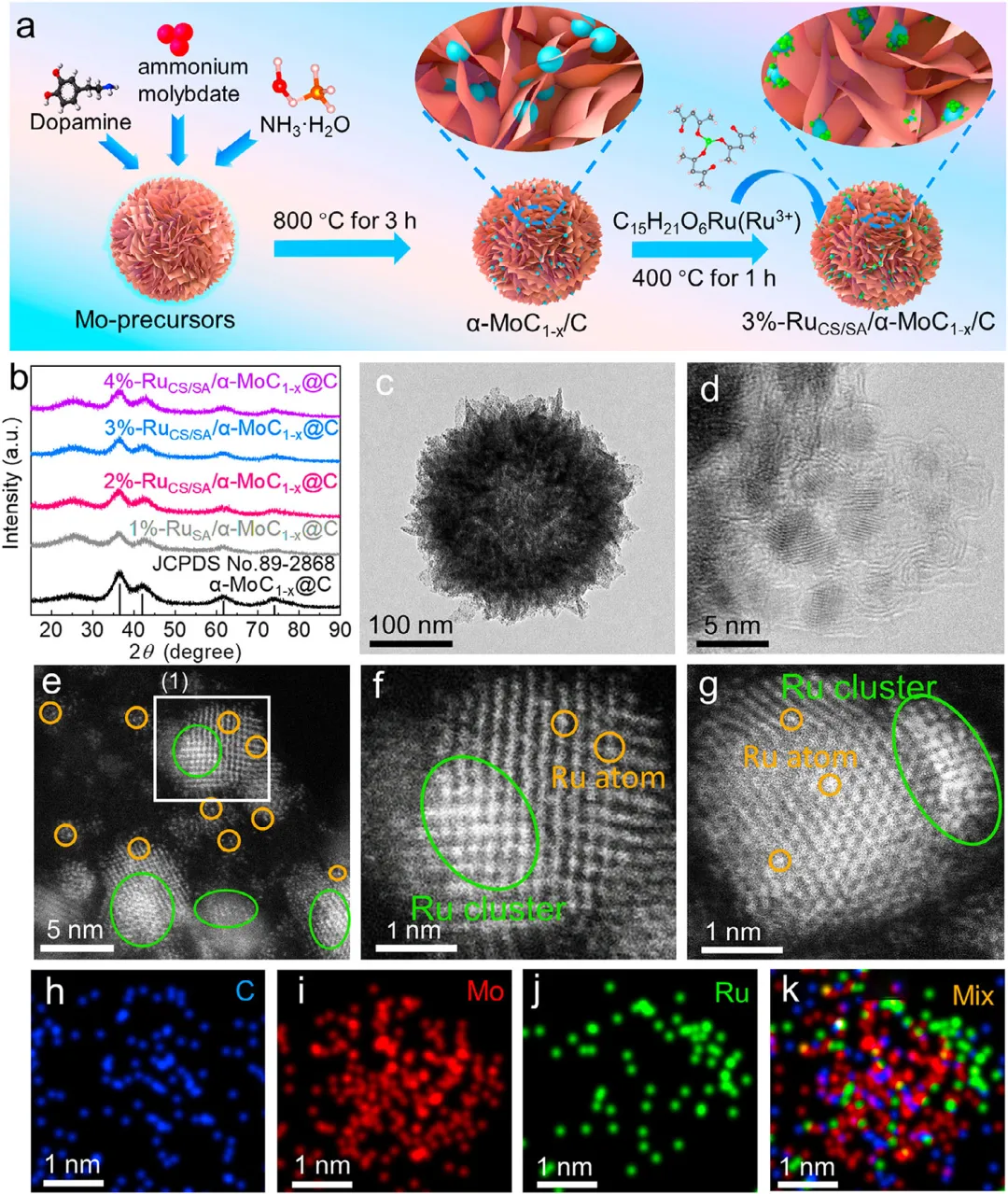
图2:a) 3%-RuCS/SA/α-MoC1-x/C的合成示意图(粉色、蓝色和绿色球分别对应C、Mo和Ru元素)。b) x%-RuCS/SA/α-MoC1-x/C的XRD图谱。c、d) α-MoC1-x/C在不同尺度下的TEM和HAADF-STEM图像。e-f) 3%-RuCS/SA/α-MoC1-x/C在不同尺度下的HAADF-STEM图像。g) 选区的电子衍射范围图像。h–j) 3%-RuCS/SA/α-MoC1-x/C的元素mapping图像和k) C、Ru和Mo的重叠元素图像。
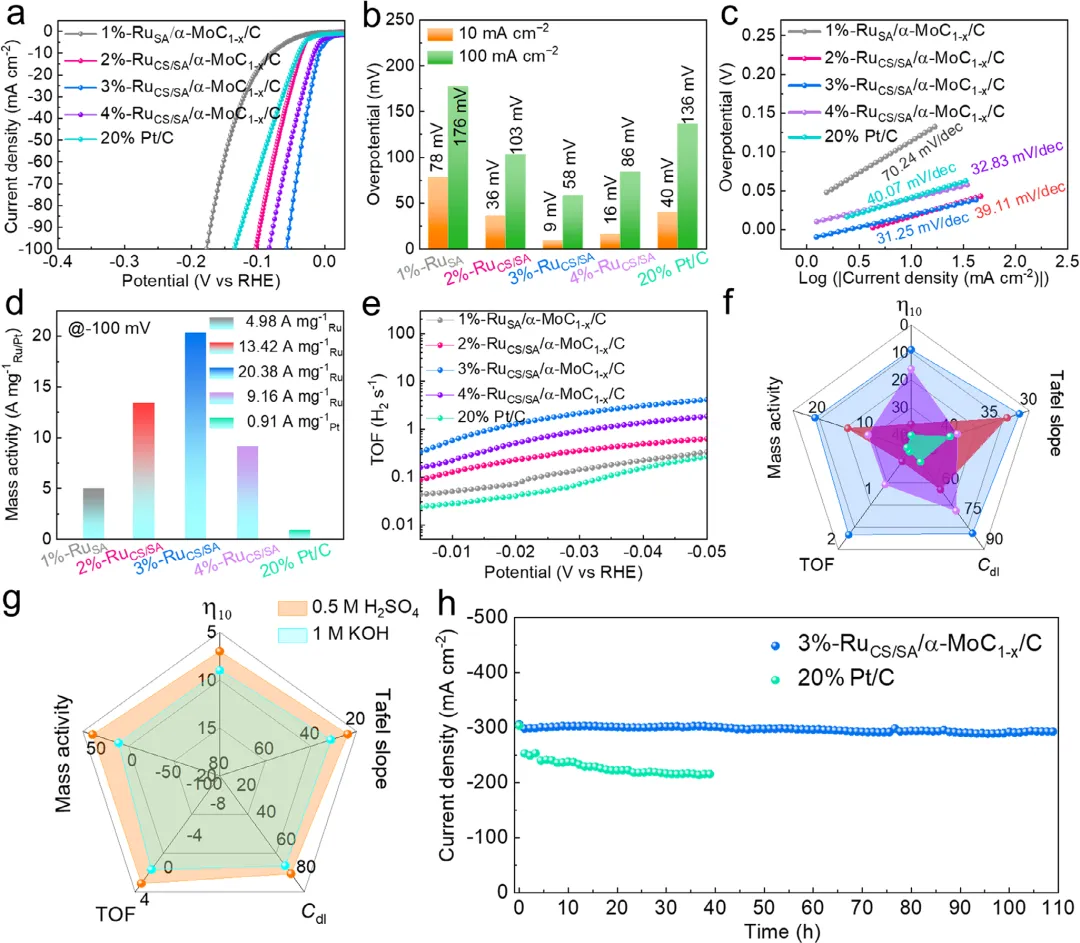
图3:a) 3%-RuCS/SA/α-MoC1-x/C与20 wt% Pt/C及其他不同Ru负载样品的极化曲线对比。b) 电流密度为10 mA cm−2和100 mA cm−2时的过电位,c) 由相应HER极化曲线得到的Tafel图。d) -100 mV(相对于RHE)下相对于参比样品的质量活度。e) 相对于参比样品的TOF曲线(不同的Ru负载)。f) x%-RuCS/SA/α-MoC1-x/C(其中x%代表2%、3%或4%的Ru负载比例)与商业Pt/C电催化剂的HER性能指标对比。g) 3%-RuCS/SA/α-MoC1-x/C在碱性电解液和酸性电解液(0.5 m H2SO4)中的HER性能指标对比。h) 3%-RuCS/SA/α-MoC1-x/C和20% Pt/C在300 mA cm−2下的计时电流测试(I–T)。
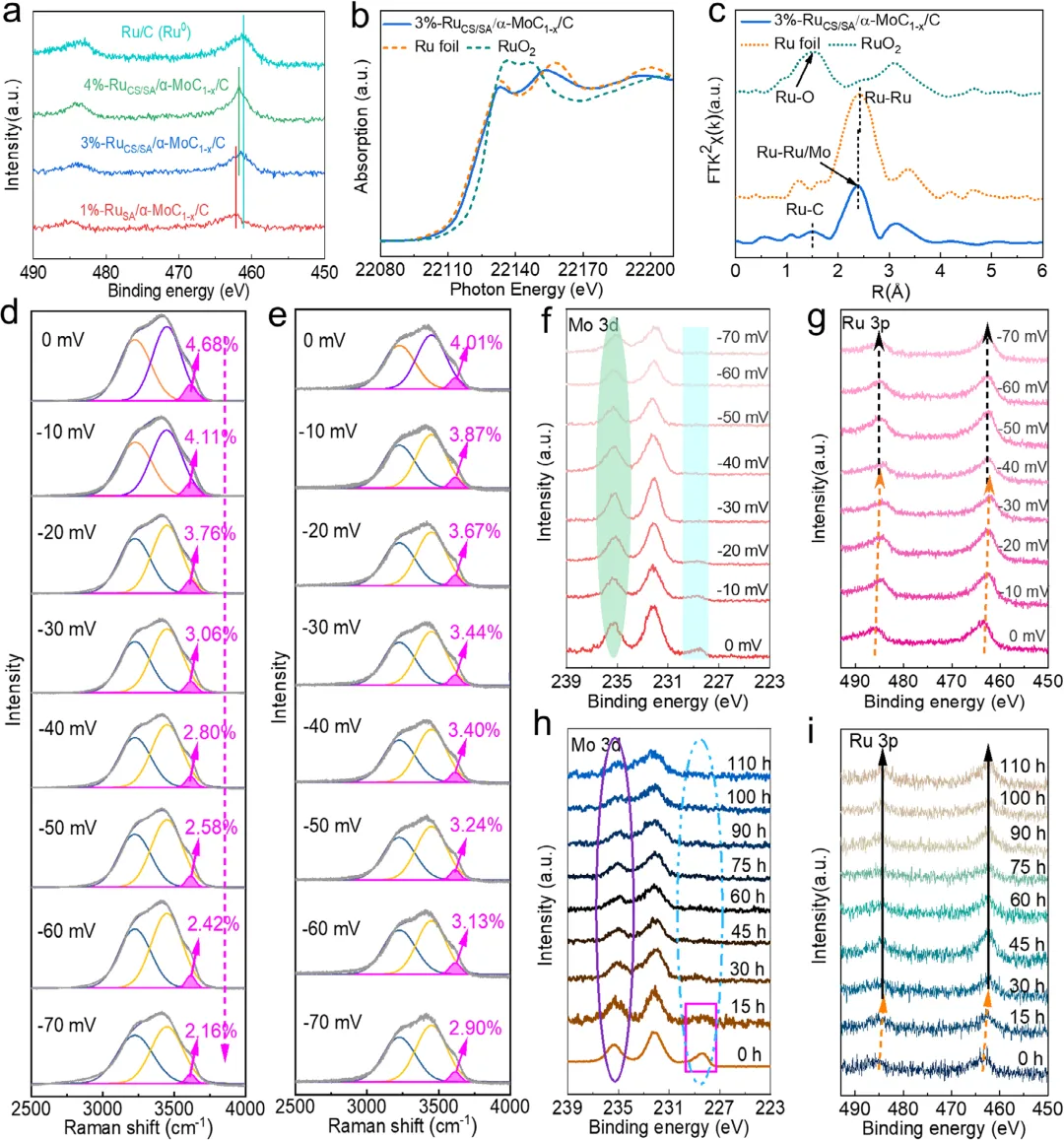
图4: a) Ru 3p谱XPS图样。b) 3%-RuCS/SA/α-MoC1-x/C、Ru箔和RuO2的Ru K边XANES谱。c) 相应的傅里叶变换Ru K边EXAFS谱。d) 1%-RuSA/α-MoC1-x/C的界面水信号的操作Raman谱。e) 3%-RuCS/SA/α-MoC1-x/C的界面水信号的操作Raman谱。f,g) 3%-RuCS/SA/α-MoC1-x/C用于HER的准原位Mo 3d和Ru 3p XPS谱分析。h,i) HER不同阶段中的Mo 3d和Ru 3p XPS谱。
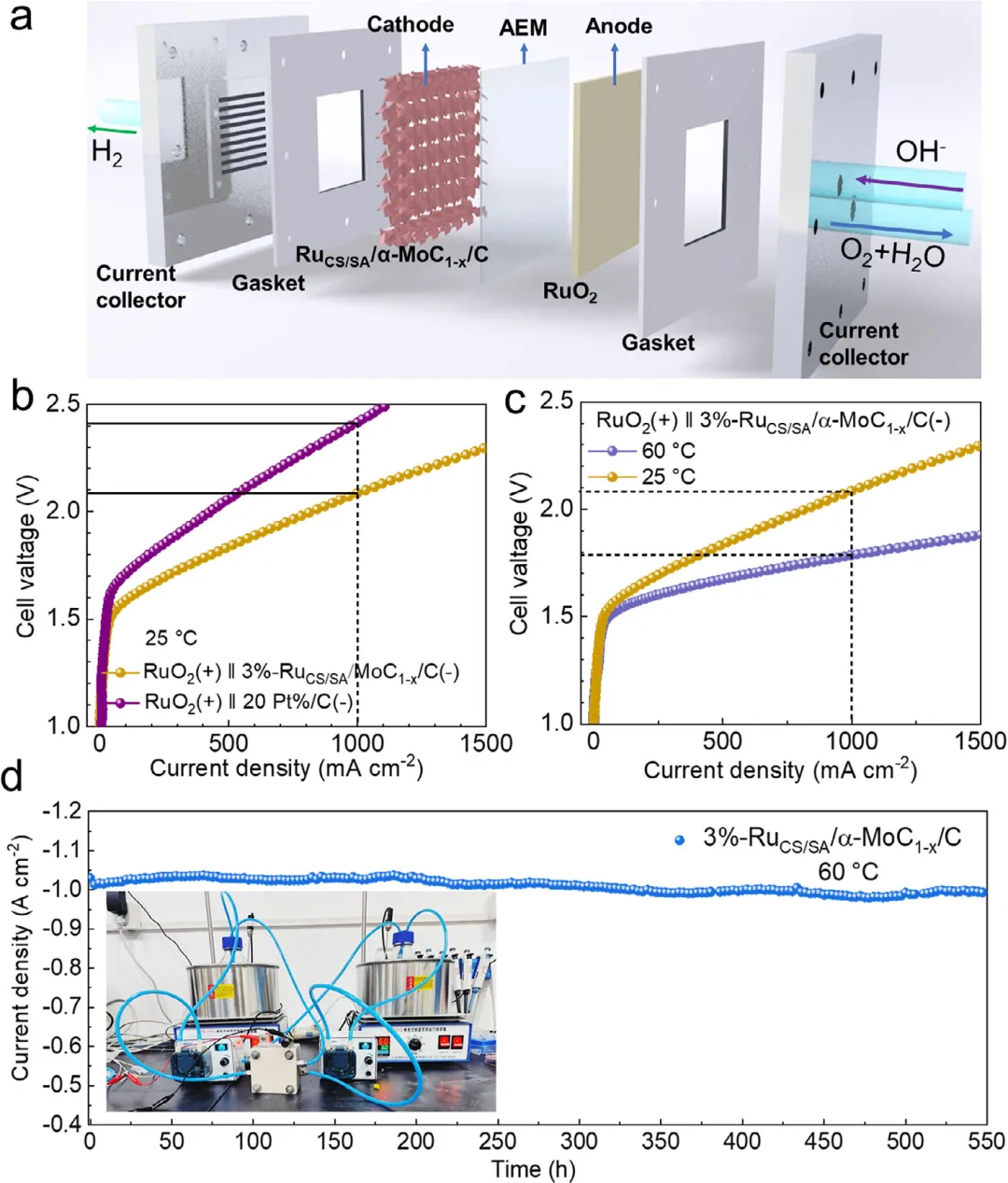
图5: a) AEMWE装置示意图及该AEMWE在碱性介质中的H2生成。b) 在25 ℃下使用3%-RuCS/SA/α-MoC1-x/C和商业20% Pt/C作为阴极催化剂的AEMWE极化曲线。c) 使用3%-RuCS/SA/α-MoC1-x/C在60 ℃和25 ℃下测量的AEMWE极化曲线。d) 基于阴离子交换膜(AEM)的电解槽中3%-RuCS/SA/α-MoC1-x/C在60 ℃下的耐久性测试。
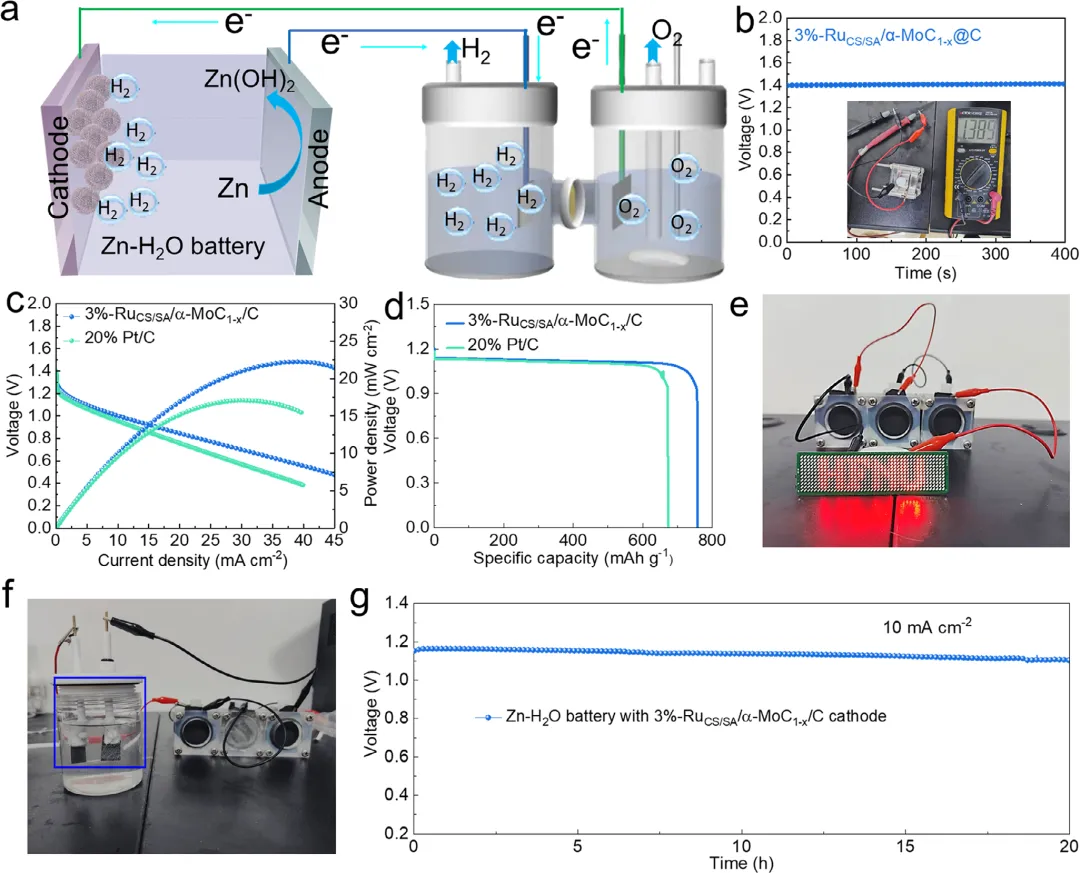
图6: a) 双Zn–H2O电池驱动电催化水分解的耦合配置示意图。b) 配备3%-RuCS/SA/α-MoC1-x/C的Zn–H2O电池的开路电位(OCP)。c) 配备3%-RuCS/SA/α-MoC1-x/C的Zn–H2O电池的LSV曲线(左侧y轴)和功率密度(右侧y轴)。d) 3%-RuCS/SA/α-MoC1-x/C和20%Pt/C的电压-比容量图。e) 由三节串联的3%-RuCS/SA/α-MoC1-x/C阴极组装的Zn–H2O电池点亮的一打LED照片。f) 由三节串联Zn–H2O燃料电池供电的水电解槽中阴极HER和阳极OER的数字照片。g) 使用Zn–H2O电池的长期耐久性测试
综上,作者通过一种简便的热解策略,构建了一种由富缺陷α-MoC1-x负载的钌团簇与相邻单原子协同催化的复合电催化剂(RuCS/SA/α-MoC1-x/C)。本工作核心在于揭示并验证了一种新颖的“电子桥接”机制:Ru单原子向富含空位的α-MoC1-x载体注入电子,而后者又将这些电子传递给邻近的钌团簇,从而实现了对不同类型钌活性位点电子结构的协同调控。这种精密的电子调制显著削弱了钌团簇对氢中间体(H*)的吸附强度,并加速了氢气的脱附动力学。
得益于此,优化后的3%-RuCS/SA/α-MoC1-x/C催化剂在碱性析氢反应(HER)中展现出卓越性能,仅需9 mV的超低过电位即可达到10 mA cm-2的电流密度,其质量活性和转换频率(TOF)均远超商用20% Pt/C催化剂。理论计算与包括原位拉曼、准原位XPS及XAFS在内的多种先进表征手段共同证实了这一独特的电荷转移路径和反应机理。
更重要的是,该催化剂在实际应用中也表现出巨大潜力,不仅在阴离子交换膜水电解(AEMWE)槽中于工业级电流密度(1.0 A cm-2)下稳定运行550小时,还能作为高效阴极驱动Zn–H2O电池,实现高功率密度(22.50 mW cm-2)和长达20小时的稳定放电。本工作为设计高效、低成本的贵金属基电催化剂提供了一条极具前景的新范式。
Precision-Engineered Electronic Modulation of Ruthenium Clusters and Single Atoms on Vacancy-Rich α-MoC1-x Enables Efficient Electrocatalytic Water Splitting. Adv. Mater., 2025. https://doi.org/10.1002/adma.202519840.
#南京大学#金钟#合肥工业大学#巢湖大学#贵金属催化剂#团簇#电子桥接#HER#工业级


