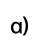论文:Angew. Chem. Int. Ed. 2025, 64, e202507569
第一作者:SongTongxin、Chenyang Shen
通讯作者:祝艳
通讯单位:南京大学
DOI:10.1002/anie.202507569
【研究背景】
1)研究对象(课题背景)
由Ti─S─金属(金属=Pd和Ag)模体组成的具有独特界面的异二聚催化剂Ag4Pd2-S-Ti4。
2)研究意义(研究此课题的必要性)
环己酮肟(CHOX)是尼龙-6的主要原料,传统合成方法面临高耗能、低选择性等挑战。电化学合成CHOX技术利用硝酸盐电还原生成关键中间体羟胺(NH2OH),再与环己酮耦合,具有绿色环保、条件温和等优势,在能源转化与精细化工领域具有重要应用价值。该研究为异相催化剂的设计提供了功能整合的创新思路,推动了原子级精准催化体系在电化学合成中的发展。
3)领域难题(该领域存在的不足与挑战)
在电还原条件下,获得高硝酸盐转化率和高环己酮肟选择性的催化剂面临挑战:一方面,硝酸盐易停留在亚硝酸盐阶段而不进一步还原;另一方面,N-O键易断裂并进一步氢化生成氨。因此,如何设计一种催化剂,既能高效还原硝酸盐生成亚硝酸盐,又能进一步将其选择性地还原为羟胺(NH2OH),并避免过度还原生成氨(NH3)或亚硝酸盐滞留,成为实现该过程的关键。
【研究现状】
1)已有哪些研究
已有研究中,用于电化学合成 CHOX 的催化剂包括Au、Ag、Cu 及其合金等金属簇,以及二氧化钛、多氧钛簇等氧化物材料。原子级精确的金属簇和多氧金属酸盐簇因可通过单晶 X 射线晶体学解析完整结构,便于识别催化活性位点和活性物种,在原子级催化体系开发中受到关注。
2)已有研究有什么不足
单一金属簇(如Ag4Pd2(SR)8)对 NO3- 还原生成 NO2-的法拉第效率较高,但 NO2- 难以进一步还原,导致 CHOX 产率低;单一多氧钛簇(如Ti4−iPrOH)对 NO2-还原具有活性,但对 NO3-还原的起始电位较负,且易生成 NH4+ 等副产物;两种簇的物理混合物催化效率极低,CHOX 的法拉第效率仅为 17.8%,无法实现功能协同。此外,传统催化剂难以同时解决硝酸盐转化率低和 CHOX 选择性差的问题,且催化机理中的中间体转化路径尚不明确。
【研究思路】
1)新概念、测试/理论/结构设计新方法
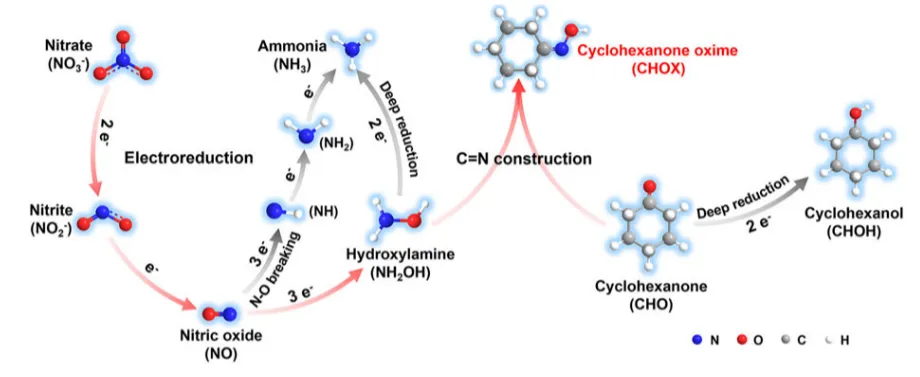
图1.以硝酸盐为氮源的环己酮肟的电化学合成示意图
提出 “异二聚簇基配对催化剂” 设计概念,通过化学键合将金属簇(Ag4Pd2(SR)8)与多氧钛簇(Ti4−iPrOH)整合,构建具有 Ti─S─Pd/Ag 界面的一体化催化体系。金属簇单元负责将 NO3-选择性脱氧生成NO2-,多氧钛簇单元负责 NO2-进一步加氢生成 NH2OH,界面结构促进中间体在两个活性位点间的高效传递,实现 “分步催化 + 协同增效”,实现了硝酸盐高效电化学还原合成CHOX,选择性高达100%,法拉第效率达68.5%。
【研究方案】
1)具体的研究方法(理论、实验)
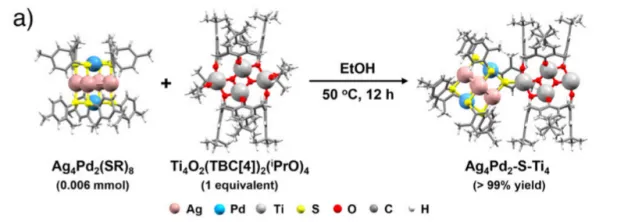
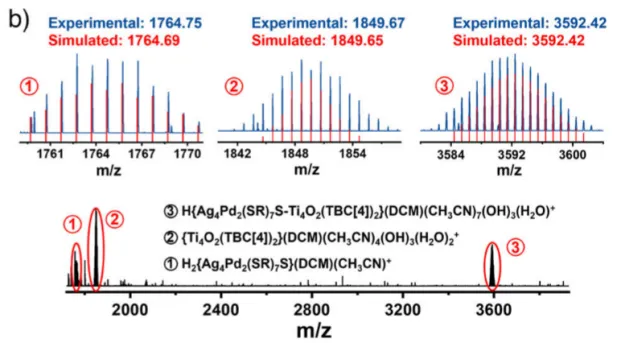
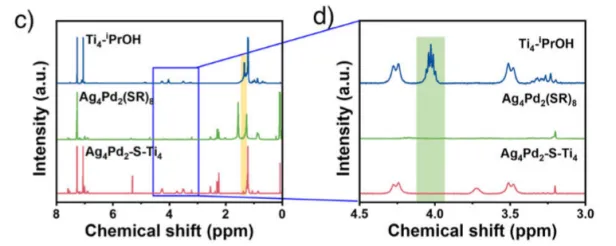
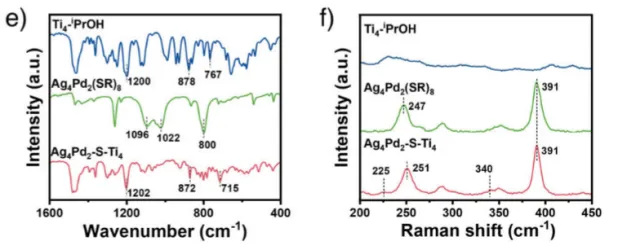
图2 : 异二聚体Ag4Pd2-S-Ti4催化剂的化学合成及组成。a) 以Ag4Pd2(SR)8和Ti4−iPrOH为前驱体,在50℃乙醇中反应12h制备Ag4Pd2-S-Ti4催化剂示意图;b) Ag4Pd2-S-Ti4样品的ESI-MS图谱;c) 三个样品的核磁共振氢谱;d) 图2c中蓝色方框的放大图;e) 傅里叶变换红外光谱;f) 拉曼光谱。
通过溶剂热反应(乙醇中50℃反应 12h)实现两个簇的共价连接:Ag4Pd2(SR)8中的 C─S 键断裂产生裸露 S 原子,Ti4−iPrOH 中的异丙氧基在乙醇中脱除形成不饱和 Ti 原子,两者通过 Ti─S 键结合,同时保留金属簇中的S─Pd/Ag 键,形成 Ti─S─金属界面。
然后通过多种谱学手段证实了催化剂的结构与界面形成。ESI-MS确认了异二聚体的分子量。¹H NMR结果表明异丙氧基信号消失,说明其已脱落。FTIR图上出现了Ti–S键振动峰(~715 cm⁻¹)。Raman图也有Ti–S键振动峰(225 和 340cm⁻¹)出现。X-射线光电子能谱 (XPS) 分析表明,Pd结合能升高,Ag结合能降低,说明电子结构改变。XANES/EXAFS:Ti价态升高,出现Ti–S配位环境。这些证据表明催化剂形成了Ti–S–Pd/Ag界面,是催化活性的关键。
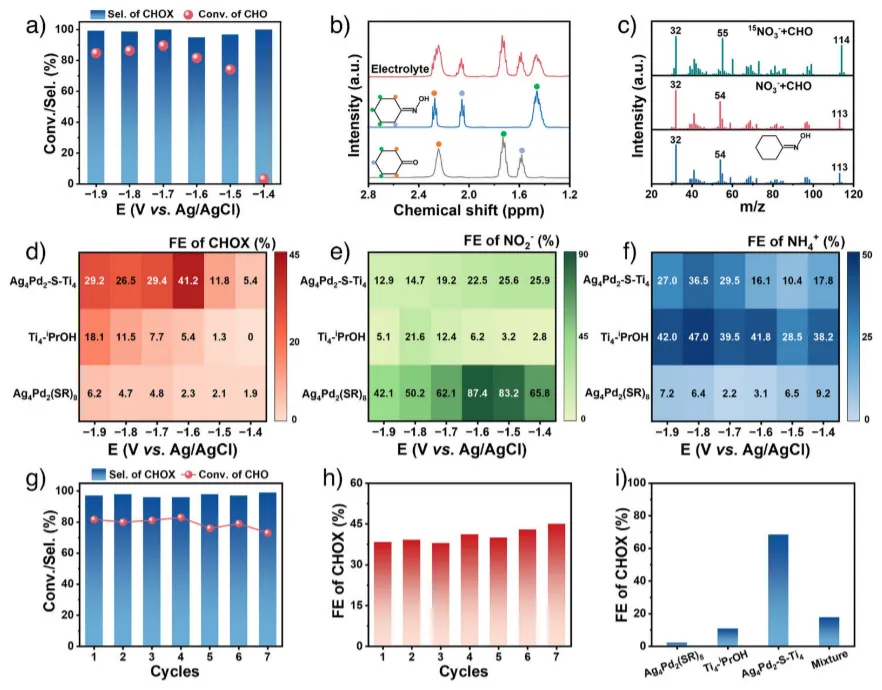
图3.a) Ag4Pd2-S-Ti4催化剂在0.4 M KNO3和0.05 M CHO溶液中的电位相关CHO转化率和CHO选择性。b) Ag4Pd2-S-Ti4催化电解液和标准CHO和CHOX样品的1H NMR。c) 在Ag4Pd2-S-Ti4催化剂作用下,CHOX标样、NO3-电解液的液体产物与CHO反应,15N标记的15NO3-电解液与CHO反应。d)-f) 在Ag4Pd2(SR)8,Ti4-iPrOH和Ag4Pd2-S-Ti4催化体系中,CHOX,NO2−和NH4+的热图。g) Ag4Pd2-S-Ti4在−1.6V相对于Ag/AgCl的电位下的可回收性。h) 在为−1.6V,Ag/AgCl七次循环实验中,CHOX在Ag4Pd2-S-Ti4上的FEs。i) 分别在Ag4Pd2(SR)8、Ti4-iPrOH、Ag4Pd2-S-Ti4以及机械混合物Ag4Pd2(SR)8和Ti4-iPrOH上 CHOX的FEs。实验是在0.5 M KNO3和0.1 M CHO的电解液中进行的。
接下来评估了该催化剂用于环己酮肟电化学合成的催化性能,在0.5 M KNO3+ 0.1 M CHO的碱性电解液中测试。图 3a 显示了Ag4Pd2-S-Ti4催化剂在不同施加电位下连续 5 小时计时电流(i-t)测试后的环己酮转化率和环己酮肟选择性。在整个电位范围内,环己酮肟的选择性为 95.0%~100%,环己酮转化率最高可达 89.7%。环己酮、环己酮肟及反应后电解质的核磁共振和质谱分析表明,反应体系中确实生成了环己酮肟(图 3b,c)。当使用15N标记的15NO3−作为氮源时,分子离子峰的质荷比为 32、55 和 114,证实合成的环己酮肟确实来源于硝酸盐(图 3c)。图 3d-f 显示了Ag4Pd2(SR)8、Ti4−iPrOH和Ag4Pd2-S-Ti4催化体系中不同产物的法拉第效率图。图 3d 中,Ag4Pd2-S-Ti4的环己酮肟法拉第效率显著高于Ag4Pd2(SR)8和Ti4−iPrOH,在-1.6 V(vs. Ag/AgCl)电位下达到 41.2%。且Ag4Pd2-S-Ti4催化剂在催化反应过程中表现出良好的稳定性,因此具有优异的可循环性和长期稳定性(图 3g,h )与预期一致,相较于Ag4Pd2(SR)8和Ti4−iPrOH的机械混合物,Ag4Pd2-S-Ti4催化剂的环己酮肟法拉第效率提高了约 4 倍(图 3i )。综合来看,Ag4Pd2-S-Ti4的催化性能优于大多数已报道的催化剂。
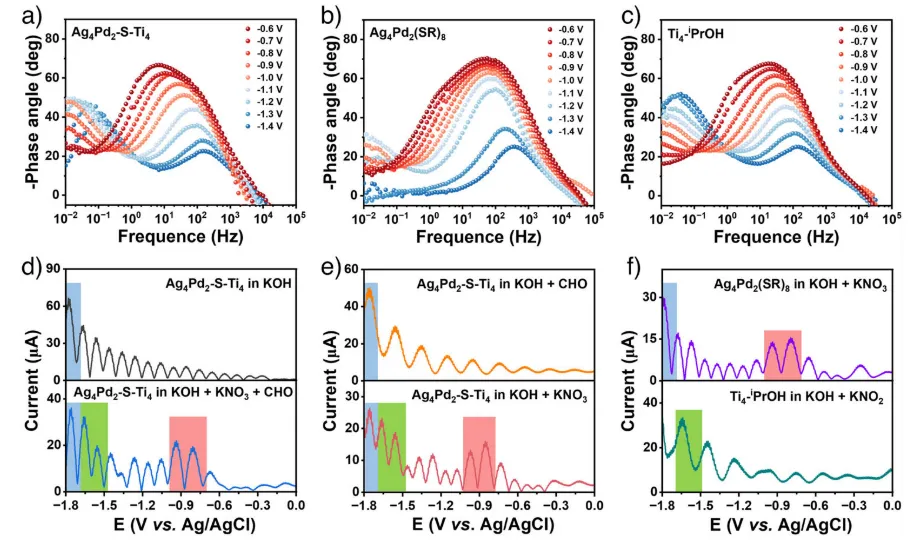
图4.三种催化剂的电化学性质和动力学行为。在含KNO3和CHO的电解液中,a)Ag4Pd2-S-Ti4,b)Ag4Pd2(SR)8和c)Ti4-iPrOH催化剂上的电位依赖的Bode相角曲线。d)-f)不同电解液体系的五次谐波FTACV曲线。频率=10Hz,幅度=0.1V。
开展机理研究,以明确Ag4Pd2-S-Ti4催化剂高性能的来源。FTACV结果显示两个还原峰,对应 NO3-到 NO2-和 NO2- 到NH2OH的两步还原,为整个催化机理提供了最关键的实验证据之一。无论是Bode图(电荷转移动力学)还是FTACV(多步电子转移),都清晰地表明Ag4Pd2-S-Ti4的性能不是两个单体功能的简单加和,而是通过界面耦合产生的1+1>2的协同效应。界面不仅连接了两个单元,更重新调控了它们的电子结构和反应路径,从而实现了高选择性的多步催化。
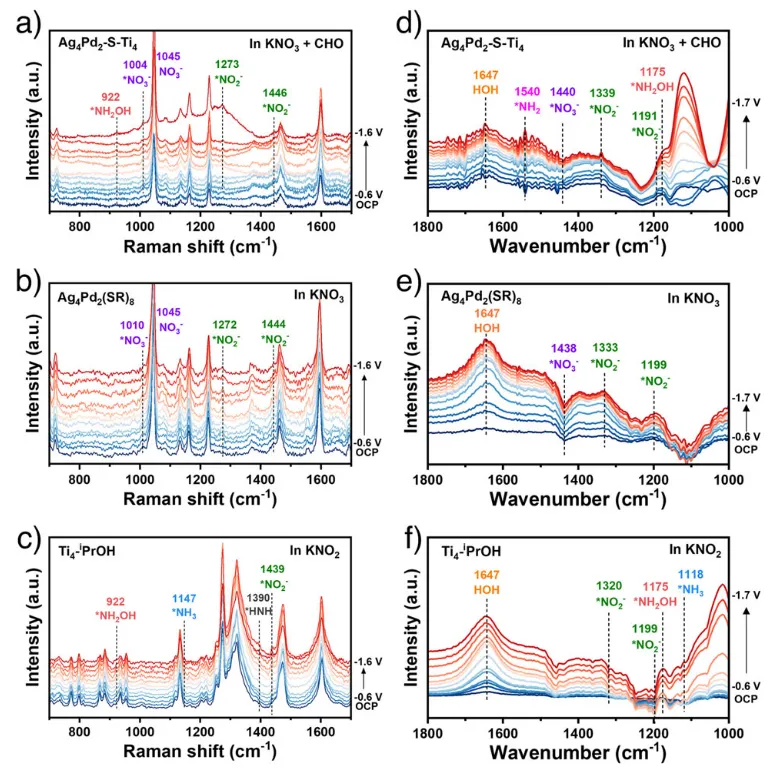
图5.与催化剂上形成的反应中间产物相对应的原位拉曼光谱和ATR-FTIR光谱。分别从a)Ag4Pd2-S-Ti4体系在含有KNO3和CHO的碱性电解液中,b)Ag4Pd2(SR)8体系在碱性KNO3溶液中,以及c)Ti4-iPrOH体系在碱性KNO2溶液中分别获得了原位拉曼光谱。在碱性电解液中,在Ag4Pd2-S-Ti4催化剂上,分别用KNO3和CHO电化学合成了CHOX;在碱性KNO2溶液中,分别在Ag4Pd2(SR)8簇上进行了NO3-的电化学还原;在碱性KNO2溶液中,分别在Ti4-iPrOH簇上进行了NO2−还原。
通过原位谱学分析(Raman、ATR-FTIR),得出Ag4Pd2-S-Ti4催化剂中Ag4Pd2(SR)8和Ti4−iPrOH的两个功能单元能够区分硝酸盐和亚硝酸盐,分别催化硝酸盐脱氧和亚硝酸盐氢化。展示了在Ag4Pd2-S-Ti4上,含氮物种沿着 NO3⁻ → NO2⁻ → NH2OH 的理想路径演进。
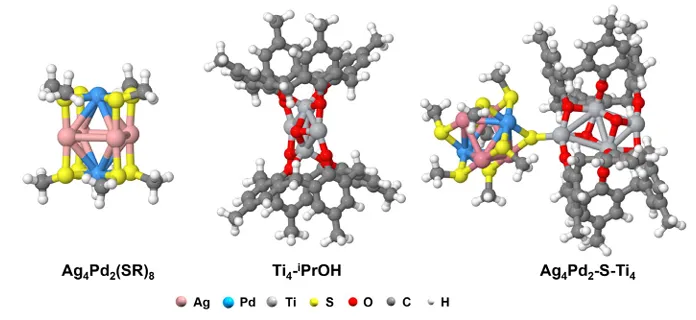
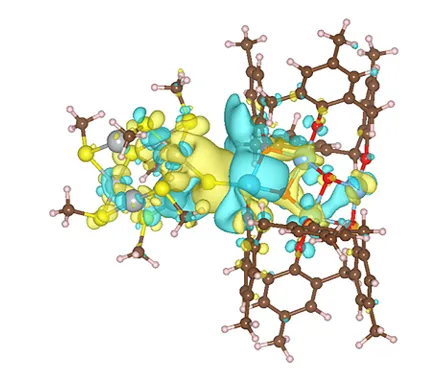
图6.a)Ag4Pd2(SR)8、Ti4-iPrOH 和 Ag4Pd2-S-Ti4 的优化几何结构;b)Ag4Pd2-S-Ti4的差分电荷密度分布。黄色和蓝色等值线分别表示电子密度的积累和减少
通过密度泛函理论(DFT)计算进一步深入研究了上述三种催化剂上硝酸盐的还原路径。将去除一个配体的Ag4Pd2(SR)8 与不含异丙氧基的Ti4−iPrOH簇耦合,构建催化剂,其优化构型如图5a所示。电子密度差图(图5b)显示,簇-簇界面存在明显的电荷转移。
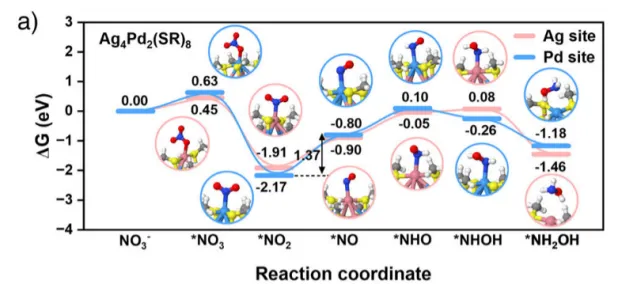
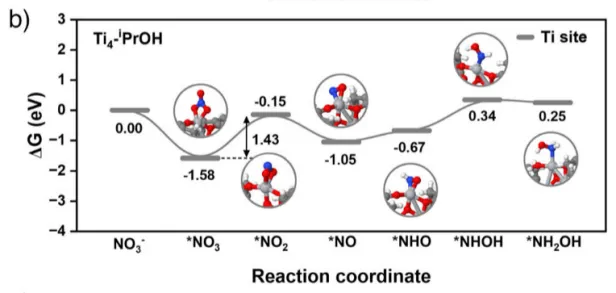
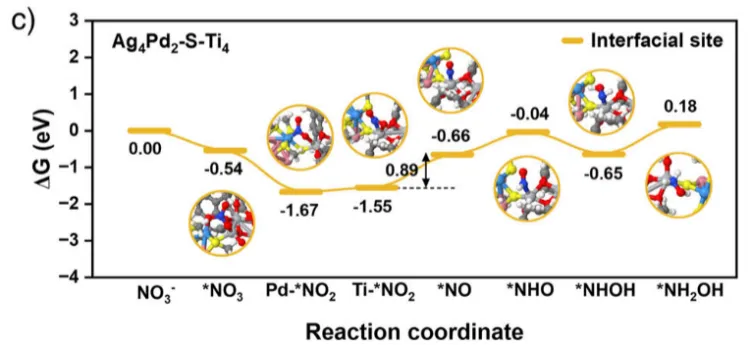
图7.三种催化剂上 NO3⁻ 还原为 NH2OH 的途径分别绘制了在a) Ag4Pd2(SR)8簇合物、b) Ti4−iPrOH簇合物和c) Ag4Pd2-S-Ti4催化剂上NO3-转化为NH2OH的吉布斯自由能图。插图显示了反应过程中各反应中间体的优化构型。蓝、粉、黄、红、灰、白、深蓝和深灰的球分别代表钯、银、硫、氧、钛、氢、氮和碳原子。
最后展示了硝酸盐还原为羟胺过程中基于吉布斯自由能变(∆G)的自由能演变及相应的吸附构型。结果表明,Ag4Pd2-S-Ti4体系中硝酸盐还原为羟胺的整个反应路径比单独的Ag4Pd2(SR)8或Ti4−iPrOH更顺畅,相应的决速步为*NO2还原为*NO,其吉布斯自由能值更低,仅为 0.89 eV。研究进一步阐明了与两个功能单元相连的界面 Ti─S─金属位点在关键羟胺物种合成中的独特作用,其中每个单元都能为硝酸盐源的还原过程发挥各自的作用。
【研究结果】
1)发现什么或得到什么结论
本研究成功制备了一种异二聚簇基催化剂 Ag4Pd2-S-Ti4 ,用于电化学还原合成环己酮肟(CHOX),通过化学键合将金属簇(Ag4Pd2(SR)8)与多氧钛簇(Ti4−iPrOH)整合,构建具有 Ti─S─Pd/Ag 界面的一体化催化体系。在碱性电解质中,Ag4Pd2-S-Ti4催化合成 CHOX 的选择性达 95.0%~100%,环己酮转化率最高为 89.7%,CHOX 的法拉第效率最高达 68.5%,远超单一簇(Ag4Pd2(SR)8的 FE 仅为 1.9%~6.2%,Ti4−iPrOH 的 FE 为 1.3%~18.1%)和物理混合物(FE=17.8%),性能优于多数已报道催化剂,并表现出良好的稳定性。
2)上述发现点与结论的科学意义
该研究首次通过簇-簇共价连接构建了具有明确界面结构的异二聚配对催化剂,实现了 “脱氧 - 加氢 - 缩合” 多步反应的高效协同,为解决电化学合成中底物转化率与产物选择性难以兼顾的难题提供了新思路。