南京大学国家级青年人才携手南工大副处长&国家优青Nat Catal-合成生物学-光生物催化自由基重定位实现远程C–C/C–H键的不对称酰化
- 2026-05-28 11:48:34
(点击页面左下角阅读原文,直达文献页面)
一、中文标题
光生物催化自由基重定位实现远程C–C/C–H键的不对称酰化(Photobiocatalytic radical repositioning for enantioselective acylation of remote C–C/C–H bonds)
二、发表单位及通讯作者
发表单位:南京大学(化学化工学院、配位化学国家重点实验室)、南京工业大学、南京林业大学
通讯作者:Ling Jiang(jiangling@njtech.edu.cn)Xiaoqiang Huang(huangx513@nju.edu.cn)
三、科学问题
如何在生物催化体系中实现自由基重定位,并控制重定位后产生的潜手性碳自由基的立体化学,从而实现对远程惰性C–C和C–H键的高对映选择性酰化?
四、发表时间
时间:2025年11月3日
链接:https://doi.org/10.1038/s41929-025-01435-1

五、摘要
本研究开发了一种可见光促进的硫胺素依赖性光生物催化体系,利用氮中心自由基触发自由基重定位,实现了远程C–C/C–H键的不对称酰化反应。该体系通过单电子转移过程生成氮中心自由基,后者经由C–C断裂或1,5-氢原子转移重定位至远离氮原子的潜手性碳自由基,并在酶活性位点内与酰基自由基发生对映选择性偶联。共合成43个具有δ‑、ε‑、ζ‑或η‑位羰基的手性腈类和酰胺类化合物,对映体比例最高达99.5:0.5。该策略首次将自由基重定位与酶催化结合,为远程惰性键的精准官能团化提供了新途径。
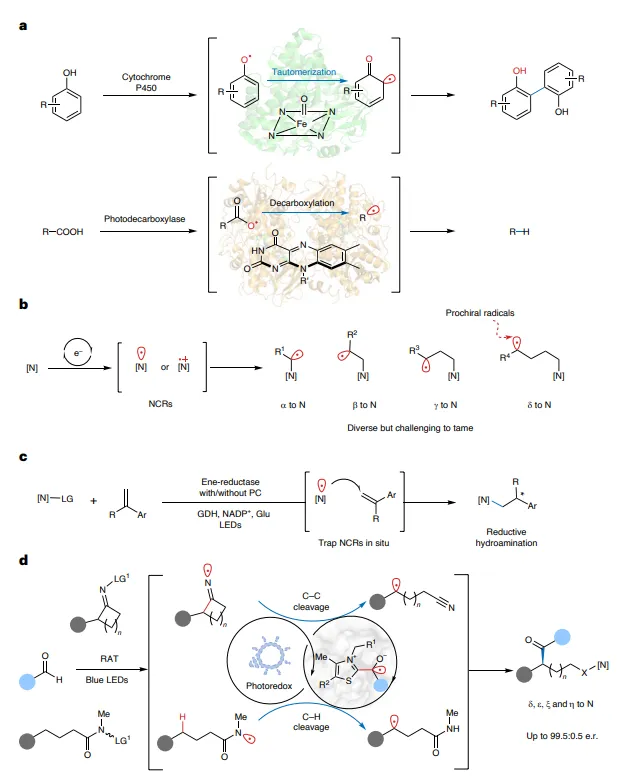
图1 光催化体系
六、研究背景
自由基反应在有机合成中具有独特优势,能实现传统离子型反应难以触及的化学转化。自由基重定位是指未配对电子通过氢原子转移、基团迁移或断裂等方式在分子内迁移,从而激活远程惰性C–H或C–C键。在天然生物体系中,自由基重定位现象较为罕见,已知案例如细胞色素P450催化的氧化C–C偶联和光脱羧酶催化的脂肪酸脱羧,但这些过程通常仅限于近程重定位,且缺乏立体化学控制。
氮中心自由基是一类重要的反应中间体,近年来在可见光催化下可通过1,2-至1,n-迁移实现远程官能团化。然而,如何控制重定位后生成的潜手性碳自由基的立体化学仍是一个巨大挑战。目前仅有少数手性铜催化剂能实现对这类自由基的对映控制,而生物催化体系在此领域的应用几乎空白。近年来,光生物催化快速发展,氮中心自由基与酶的兼容性已得到初步验证,但其反应模式多局限于自由基直接捕获,尚未涉及惰性键的激活与远程迁移。
因此,开发一种兼具自由基重定位能力和立体控制功能的生物催化体系,对于实现远程C–C/C–H键的高选择性官能团化具有重要意义。本研究旨在将光催化生成的氮中心自由基与硫胺素依赖性酶相结合,通过协同催化实现远程自由基酰化,并借助酶的活性口袋控制反应的立体选择性。
七、研究结果
结果1、C–C键自由基酰化反应的发展与优化
以4-氯苯甲醛为酰基供体、2-苯基环丁酮肟醚为自由基前体,在硫胺素依赖性酶PfBAL‑T481L‑A480G催化下,经过条件优化,在无外加光催化剂、蓝光照射、TEOA缓冲液(pH 8.5)中,实现了C–C键的断裂与远程酰化,以79%收率和97:3 e.r.获得产物。研究发现,光照射能显著提升反应效率(暗态下仅34%收率),而酶活性口袋对反应的对映选择性起关键作用。
结果2、C–C酰化反应的底物适用范围
在最优条件下,考察了苯甲醛与多种肟醚的兼容性。供电子或吸电子取代的苯甲醛均可参与反应,其中吸电子基团表现更优(如‑CF₃、卤素),产物收率18–91%,e.r.最高达99:1。环丁酮、环戊酮及环己酮肟醚均能顺利发生自由基迁移(分别对应δ‑、ε‑、ζ‑位酰化),收率良好,立体控制优异(最高99:1 e.r.),表明酶能精准识别不同距离的潜手性自由基。
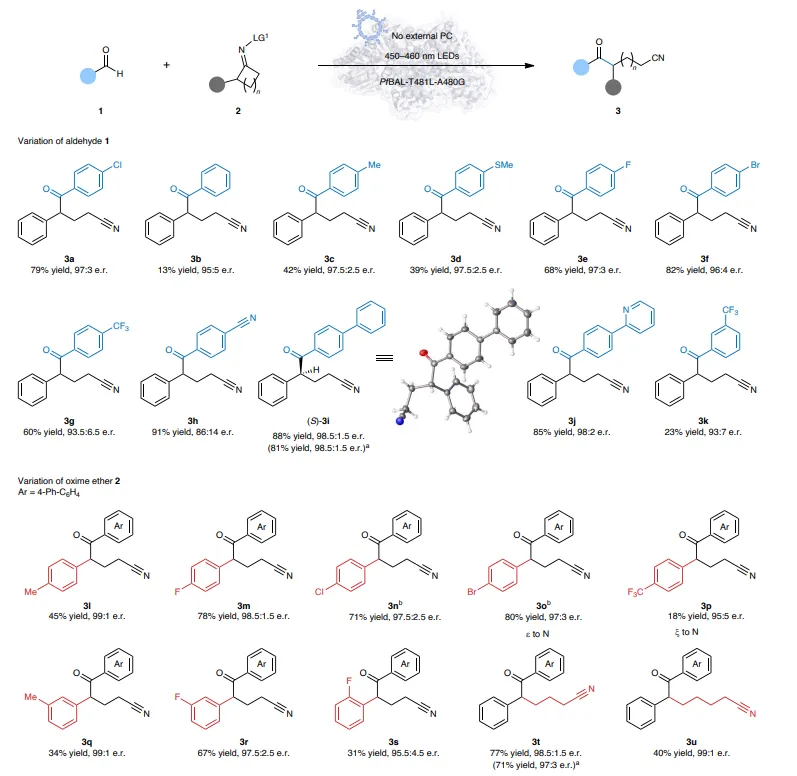
图2 底物谱
结果3、C–H键自由基酰化反应的发展与优化
以N‑ODNP酰胺为底物,通过引入天然辅因子FAD作为光敏剂,在PfBAL‑T481L‑A480G‑N283F催化下,实现了远程C(sp³)–H键的酰化。经优化,在TEOA缓冲液、蓝光照射下,获得54%收率、98:2 e.r.。光照射为本反应必需条件,凸显其与C–C酰化在引发机制上的差异。
多种苯甲醛及N‑ODNP酰胺均能顺利参与反应,包括含卤素、烷基、三氟甲基等取代的底物,收率18–85%,e.r.普遍高于95:5,最高达99.5:0.5。值得注意的是,当使用N‑甲基‑3‑苯基丁酰胺时,意外发生了超远程芳环C–H酰化(η‑位),收率85%,展现了该体系在远程官能团化中的潜在适用性。
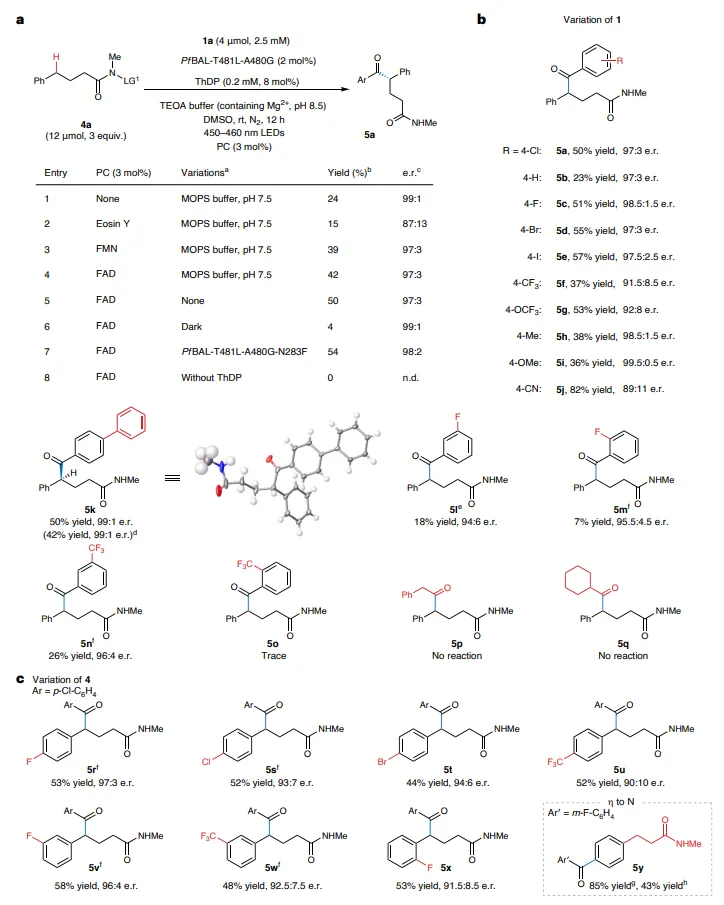
图3 条件和范围
结果4、机理研究
通过自由基捕获实验(TEMPO、1,1‑二苯乙烯)证实了自由基中间体的存在。紫外‑可见吸收与荧光猝灭实验表明,在C–C酰化中,原位生成的2,4‑二硝基苯酚可能作为内源性光敏剂;而在C–H酰化中,FAD通过还原猝灭路径促进单电子转移。分子动力学模拟显示,酶活性口袋中ThDP衍生的酰基自由基与迁移后的碳自由基距离约3 Å,且生成S构型产物的结合能更低,与实验观察的对映选择性一致。
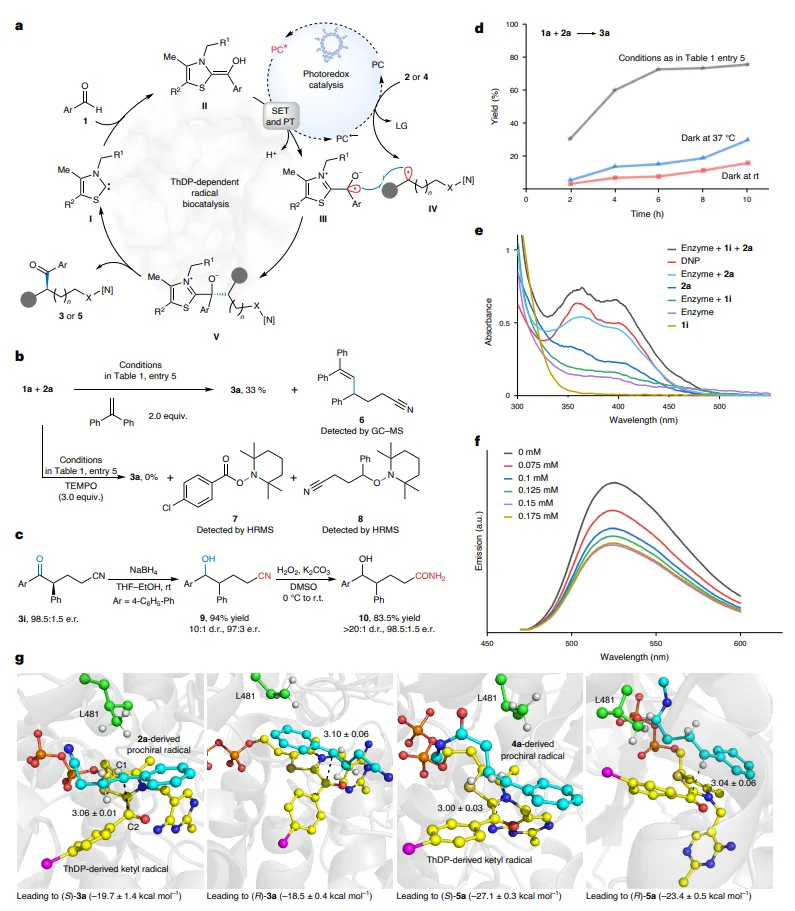
八、讨论
本研究首次将自由基重定位概念引入酶催化体系,实现了远程C–C/C–H键的高对映选择性酰化,突破了传统生物催化在惰性键活化与立体控制方面的局限。该体系在温和、水相、无金属无配体条件下运行,兼具原子经济性与立体精准性,为复杂手性分子的合成提供了新工具。未来可通过酶工程进一步拓展底物范围、提升催化效率,并探索其他类型的自由基迁移模式。该工作彰显了光‑酶协同催化在实现非天然自由基转化中的强大潜力,为绿色合成与生物催化交叉领域的发展提供了新思路。

