南京理工大学付永胜教授/朱俊武教授等人EES:纳米框架中自发磁场增强实现高效氧还原反应
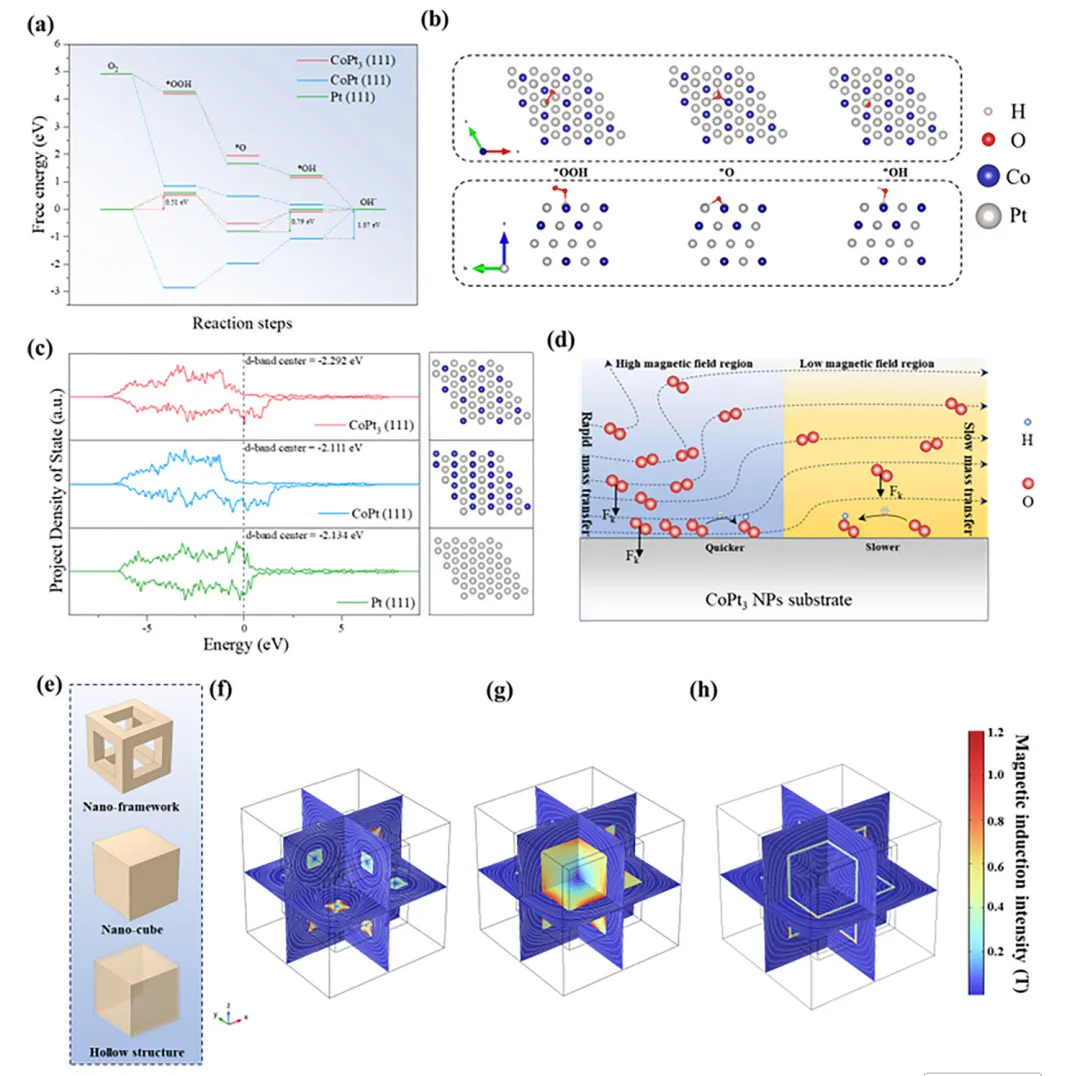
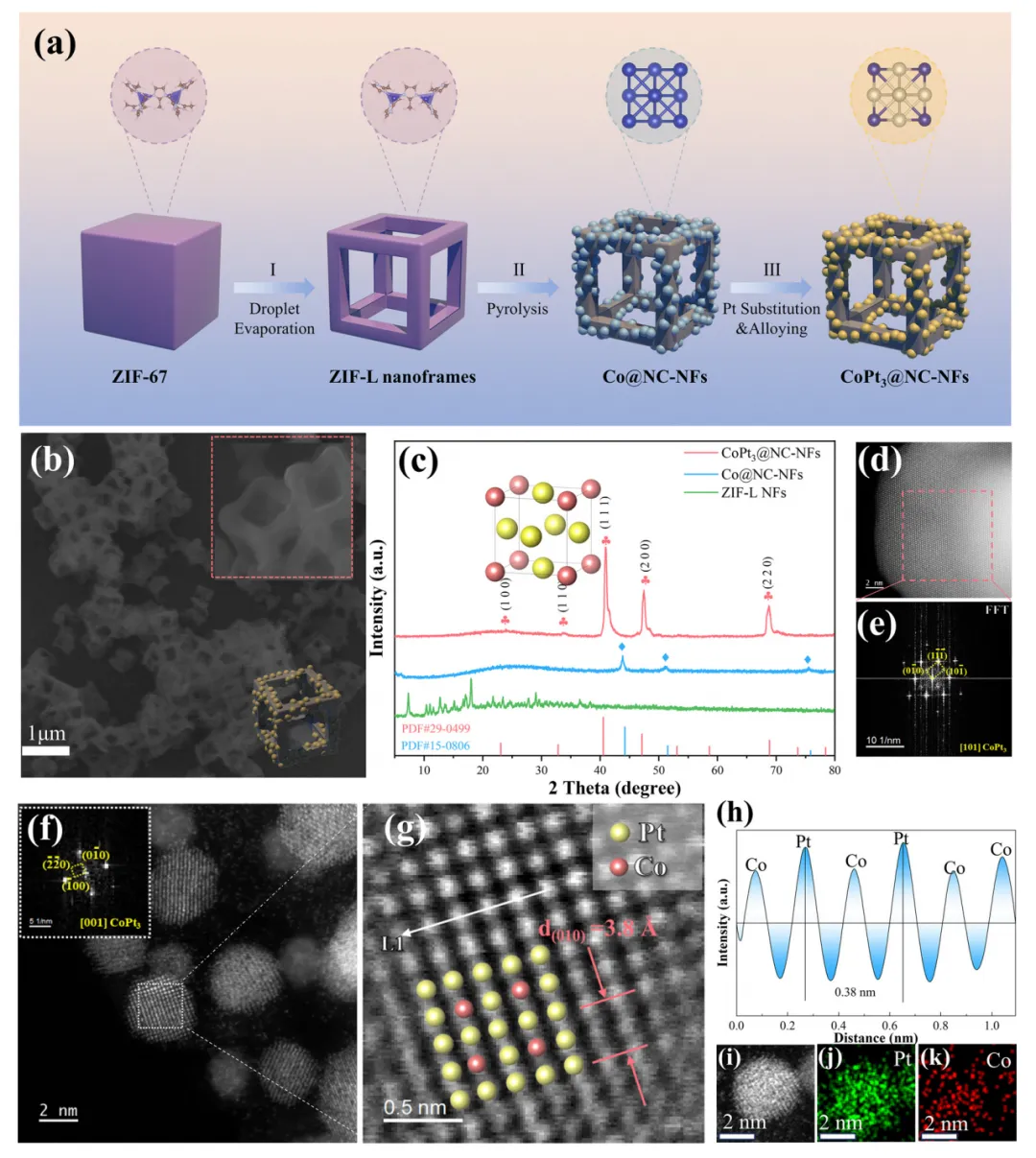
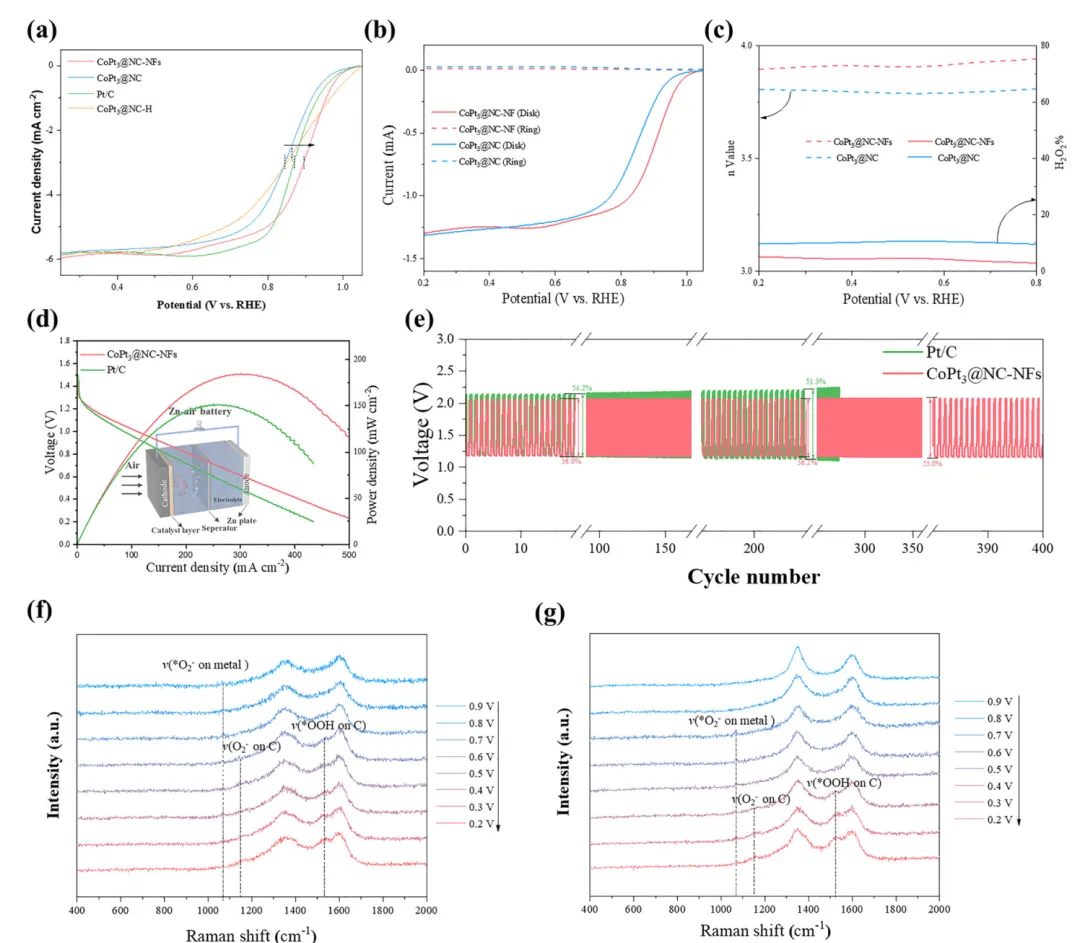
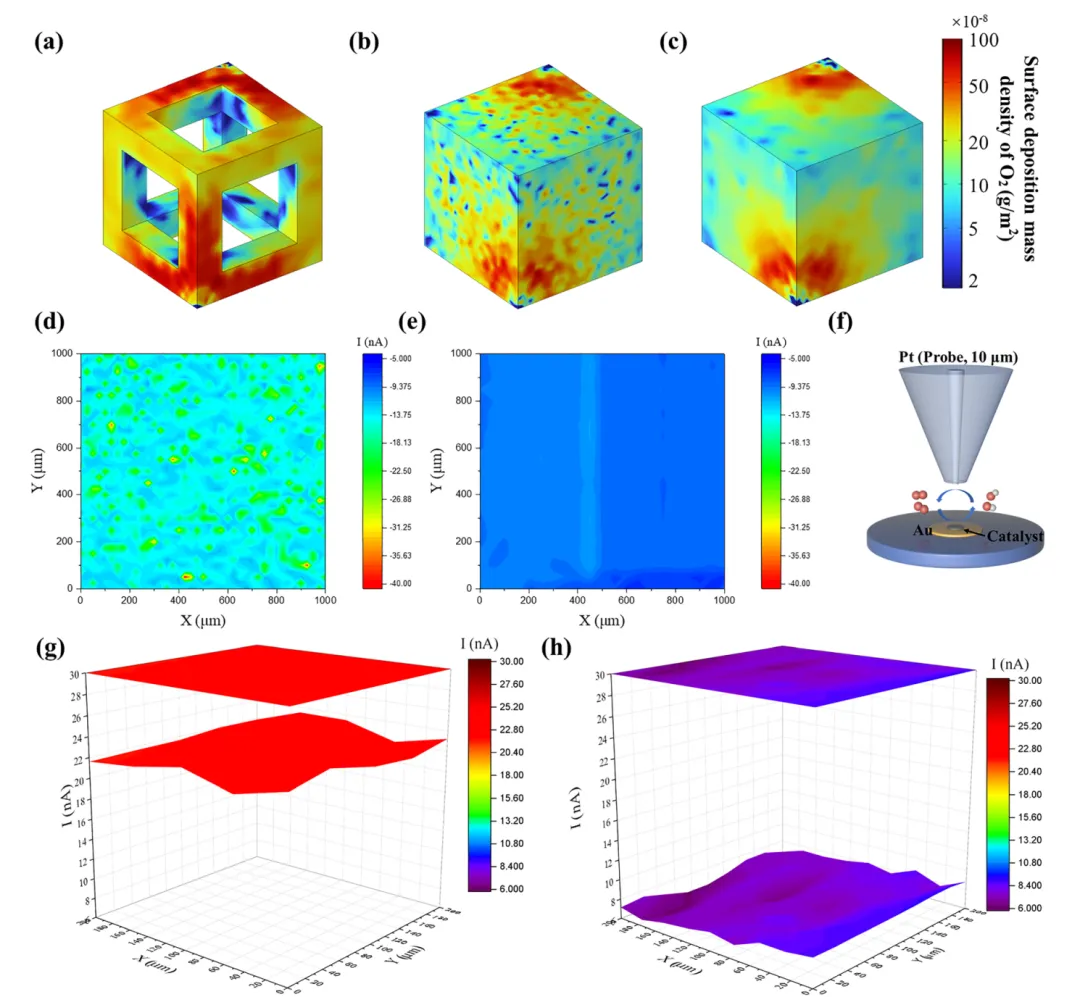
氧还原反应(ORR)是燃料电池、金属-空气电池等可持续能源转化存储器件的关键过程,但其效率受限于缓慢的O₂传质。本文制备了仅由边、角构成的CoPt₃基纳米框架催化剂(CoPt₃@NC-NFs),该结构在电化学反应中聚集电流密度,放大表面磁场强度,增强作用于顺磁性O₂分子的开尔文力,驱动O₂定向快速向催化剂高场区域迁移,提升O₂传质效率。该催化剂在0.1 M KOH中展现优异ORR性能,起始电位1.02 V、半波电位0.89 V、极限电流密度-5.8 mA cm⁻²,组装的锌-空气电池在305 mA cm⁻²下功率密度达184.8 mW cm⁻²。本研究建立了催化剂微观结构与表面磁场的关联,揭示自发磁场增强机制,为提升电催化活性提供了新的物理视角,该催化剂在新能源转化器件中具有极高的实际应用潜力。(498字)氧还原反应(ORR)作为燃料电池、金属-空气电池等能源转化技术的核心阴极反应,其效率直接决定器件整体性能,却受四电子转移的多步反应机制和缓慢的O₂传质过程制约,成为器件性能提升的关键瓶颈。近年来,科研人员围绕电子结构工程开发了杂原子掺杂、缺陷工程、纳米限域效应等策略,有效调控催化活性位点的d带中心与电荷分布,提升了ORR催化活性和反应动力学,但这类策略仍聚焦于催化活性强化,对O₂传质动力学这一核心瓶颈的解决进展有限,导致活性位点利用率低,催化潜力无法充分发挥。电催化体系中,催化剂-电解质界面传质由浓度扩散、电场迁移和对流共同调控,现有研究主要通过构筑中空、多孔结构等微纳结构工程提升浓度扩散,以强化O₂传质。但反应过程中局部浓度梯度会逐渐衰减,扩散驱动的传质效率下降,且扩散的随机性无法实现O₂在催化活性中心的定向富集,造成传质与反应位点的空间失配,大量活性位点闲置。基态O₂分子具有本征顺磁性,为磁场调控传质提供了物理基础,顺磁性物种在非均匀磁场中会受到开尔文力作用向高磁场区域迁移,但其单分子受力较弱,需结合其他传质方式发挥作用。现有研究多依赖外加磁场构建磁场梯度,却无法实现O₂在微纳尺度活性位点的定向富集。而电化学反应中电流通过材料会产生非均匀本征磁场,其强度与表面电流密度线性相关,通过结构设计聚集电流密度、增强本征磁场,成为定向引导O₂至活性位点的全新思路,相关研究目前仍处于空白阶段。(986字)本文开发了简易高效的自发表面磁场增强策略,构筑了仅由边、角构成的CoPt₃基纳米框架催化剂(CoPt₃@NC-NFs),实现O₂分子在催化活性位点的主动定向富集,显著提升ORR催化性能。有限元模拟证实,该纳米框架在电催化过程中使电流沿边界空间限域分布,局部电流密度增强并诱导自发表面磁场提升,强化的磁场进一步增大作用于O₂的开尔文力,推动O₂快速定向向催化剂表面迁移。原位拉曼光谱和扫描电化学显微镜(SECM)验证了磁场增强可加速O₂在催化剂表面的定向富集,大幅提升ORR性能。本研究阐明了纳米框架结构诱导自发磁场增强的特性,结合模拟与实验揭示了磁场增强驱动O₂在活性位点主动定向聚集的核心机制。(489字)对Pt(111)、CoPt(111)、CoPt₃(111)进行DFT计算,发现U=1.23 V时CoPt₃的决速步为*OOH形成(ΔG=0.51 eV),能垒远低于Pt和CoPt,展现最优ORR催化潜力;其d带中心更低,与氧的成键作用较弱,需强化O₂吸附以提升反应性。有限元模拟构建立方、中空、框架三种500×500×500 nm的CoPt₃模型,发现框架模型在电化学过程中产生的磁场显著强于其他结构,10 nA下其最大磁场强度为立方模型的1.25倍、中空模型的1.34倍,且磁场强度随电流增大的增强效应更显著,磁场线向催化剂表面聚集,为O₂定向迁移提供了梯度条件。以立方ZIF-67为前驱体,经马兰戈尼效应制备ZIF-L纳米框架,再经N₂退火、Pt²⁺交换与重组,成功制备CoPt₃@NC-NFs,同时制备立方结构CoPt₃@NC和中空结构CoPt₃@NC-H作对比。表征显示CoPt₃@NC-NFs保持规整纳米框架结构,CoPt₃为FCC晶型,金属颗粒粒径5~20 nm,Co、Pt、N、C元素分布均匀,形成有序金属相间结构。该催化剂石墨化程度更高、电导率更优,介孔为主且平均孔径大于6 nm,BET比表面积达180.0 m² g⁻¹,为反应提供充足活性位点与传质通道;XPS证实Pt/Co原子比接近3,且元素结合能存在轻微负移;磁滞回线测试表明其矫顽力达300 Oe,铁磁性远优于CoPt₃@NC,增强了与顺磁性O₂的相互作用。在O₂饱和0.1 M KOH中,CoPt₃@NC-NFs的ORR峰电位达0.92 V,1600 rpm下起始电位1.02 V、半波电位0.89 V,性能优于CoPt₃@NC、CoPt₃@NC-H和商业Pt/C。Koutecky-Levich曲线与RRDE测试表明,该催化剂平均电子转移数约3.96,遵循四电子转移路径,过氧化氢产率仅4.8%,催化选择性最优;12 h耐久性测试无明显电流衰减,稳定性优异。将其组装为锌-空气电池阴极,开路电压稳定在1.50 V,305 mA cm⁻²下功率密度达184.8 mW cm⁻²,远高于Pt/C基电池;不同电流密度下放电平台更高,10 mA cm⁻²下比容量达815.2 mA h g⁻¹;400次充放电循环后电压间隙无明显增大,往返效率略有提升,而Pt/C基电池300次循环后即失活,体现了该催化剂优异的实际应用性能。原位拉曼光谱监测发现,CoPt₃@NC-NFs在0.9 V即出现金属位点O₂⁻吸附峰,0.6 V出现碳位点O₂⁻和OOH峰,均远早于CoPt₃@NC,表明磁场增强加速了O₂吸附与决速步OOH的形成。有限元模拟O₂分布发现,有电流时框架模型表面O₂扩散速率与沉积量均高于立方、中空模型,1 h后O₂沉积量达9.3×10⁻⁷ mg cm⁻²,而无电流时中空结构O₂吸附能力更优,证实框架结构的磁场是O₂定向传质的关键驱动力。SECM测试显示,CoPt₃@NC-NFs的电流分布更均匀、反应区域电流值更高,TG/SC模式下其基底电流均高于20 nA,远高于CoPt₃@NC的10 nA左右,证明该催化剂能快速捕获O₂并促进其转化,验证了纳米框架诱导的表面磁场增强可有效推动O₂的快速、主动、定向传质。(1965字)本文以ZIF-L框架为前驱体,成功制备了CoPt₃纳米颗粒负载的氮掺杂碳纳米框架催化剂CoPt₃@NC-NFs。该独特的纳米框架结构在电化学过程中产生的表面磁场远强于立方结构,显著增强了作用于顺磁性O₂的开尔文力,实现O₂分子向催化剂表面的主动定向富集,提升了O₂吸附能力并促进ORR决速步*OOH的形成。该催化剂在0.1 M KOH中表现出卓越的ORR催化性能,起始电位1.02 V、半波电位0.89 V、极限电流密度-5.8 mA cm⁻²,性能优于立方结构催化剂与商业Pt/C。以其为阴极组装的碱性锌-空气电池开路电压达1.50 V,峰值功率密度184.8 mW cm⁻²,且具备优异的循环稳定性。本研究为设计高效ORR催化剂提供了重要策略,揭示了磁场在ORR中的增强机制,为深入探索材料磁效应在ORR过程中的作用提供了建设性思路。(492字)图1、(a)几种铂基材料在0 V和1.23 V下氧还原反应的吉布斯自由能图;(b)氧还原反应过程中CoPt₃(111)的优化结构;(c)CoPt₃(111)、CoPt(111)和Pt(111)的分波态密度;(d)高、低磁场区域中氧气和*OOH受到开尔文力的示意图;(e)有限元模拟中的框架、立方体、中空结构模型;(f、g、h)框架模型、立方体模型、中空模型在电化学过程中的模拟磁场分布图;图2、(a)CoPt₃@NC-NFs的合成示意图;(b)CoPt₃@NC-NFs的扫描电镜图像;(c)ZIF-L纳米框架及其衍生物的X射线衍射图谱;(d)沿[101]方向的高角环形暗场扫描透射电镜图像;(e)CoPt₃@NC-NFs纳米晶体红色选区的快速傅里叶变换图谱;(f)沿[001]方向的CoPt₃@NC-NFs高角环形暗场扫描透射电镜图像;(g)(f)中白色矩形区域的放大高角环形暗场扫描透射电镜图像及原子排列;(h)沿L1选区的对应线强度分布;(i-k)单个Pt₃Co纳米颗粒的高角环形暗场扫描透射电镜图像及元素分布;图3、(a)多种CoPt₃基催化剂和铂碳催化剂在1600转每分钟下的线性扫描伏安曲线;(b)1600转每分钟下CoPt₃基催化剂的旋转环盘电极线性扫描伏安曲线;(c)旋转环盘电极测试得到的电子转移数和过氧化氢产率;(d)CoPt₃@NC-NFs和铂碳基锌-空气电池的放电极化曲线和功率密度图;(e)CoPt₃@NC-NFs和铂碳基锌-空气电池的恒电流充放电循环曲线;(f、g)不同电势下CoPt₃@NC、CoPt₃@NC-NFs表面氧还原反应的电化学原位拉曼光谱(氧气饱和的0.1摩尔每升氢氧化钾溶液);图4、(a-c)氧气在催化剂表面的沉积分布图:框架结构、中空结构、立方结构;(d、e)1毫米×1毫米几何区域内CoPt₃@NC-NFs、CoPt₃@NC在恒高模式下的二维扫描电化学显微镜图像;(f)扫描电化学显微镜模型示意图;(g、h)0.6 V(相对于可逆氢电极)外加电势下CoPt₃@NC-NFs、CoPt₃@NC在生成/收集模式下的扫描电化学显微镜图像;Zhijie Qi, Zhenjie Lu, Pengcheng Yao, Wuxin Ba, Lianjin Wei, Duansheng Liu, Jun Jiang, Shujun Liu, Pawel J. Kulesza, Jingwen Sun, Pan Xiong, Xin Wang, Junwu Zhu, Yongsheng Fu*- Pengcheng Yao*:Department of Chemical and Biomolecular Engineering and Ralph O’Connor Sustainable Energy Institute, Johns Hopkins University, Baltimore, Maryland 21218, USA
- Junwu Zhu*:Key Laboratory for Soft Chemistry and Functional Materials, School of Chemistry and Chemical Engineering, Nanjing University of Science and Technology, Nanjing 210094, China
- Yongsheng Fu*:Key Laboratory for Soft Chemistry and Functional Materials, School of Chemistry and Chemical Engineering, Nanjing University of Science and Technology, Nanjing 210094, China
https://doi.org/10.1039/d5ee06929g本文来源各大出版社论文数据库,版权归文章出版社所有;本文内容采用AI辅助整理生成,如有错漏请私信联系;本文仅用于学术分享,转载请注明出处;如需推广本人学术成果和商务合作请私信联系,若有错漏或侵权请私信联系删除或修改!