三院院士!新加坡国立大学刘斌/何迁&南京大学姚颖方最新JACS:不对称双金属界面助力太阳能驱动CO₂还原制CH₄耦合C-H活化!
揭示用于太阳能驱动二氧化碳还原制甲烷并耦合C-H活化的非对称双金属界面https://pubs.acs.org/doi/10.1021/jacs.5c19901太阳能驱动二氧化碳与生物质衍生醇的转化,为减少二氧化碳排放并生成高附加值化学品提供了一种颇具前景的策略。然而,由于反应路径复杂且C-H键活化迟缓,在实现高效光催化二氧化碳还原制甲烷的同时,对生物质衍生醇进行增值利用仍颇具挑战。本研究中,我们报告了一种巧妙设计,即通过在富含空位的金属有机框架材料(MOFs)中锚定半导体纳米团簇,构建非对称双金属催化中心,实现了二氧化碳向甲烷的光还原与C-H氧化的串联反应。通过在富含氧空位的Mil-125(Ti)-NH₂中负载氧化铁(Fe₂O₃)团簇,界面氧空位使新形成的Fe-O键成为构建Z型机制(Z-scheme)的原子级电子转移路径。此外,界面空位引发紧密相互作用并调控d带中心,形成非对称钛-铁(Ti-Fe)双金属位点。值得注意的是,该体系实现了甲烷生成选择性高达87.0%,同时苄醇氧化为苯甲醛的选择性达100%。机理研究表明,异核Ti-Fe单元通过d-p杂化协同促进CO向CHO的加氢过程,使甲烷合成在热力学上更有利,而Fe₂O₃产生的光生空穴则优先氧化苄醇的α-C-H键。另一基于MOFs的催化剂进一步验证了这一结论。本研究为构建非对称双金属界面以同步实现选择性二氧化碳光催化甲烷化与C-H键活化提供了通用设计思路。利用太阳能将二氧化碳还原为碳氢燃料,为减少二氧化碳排放和生成高附加值化学品提供了一个可持续且极具吸引力的平台。在众多潜在太阳能燃料中,光催化二氧化碳还原制甲烷因能量密度高且与现有基础设施兼容而备受关注。然而,考虑到八电子转移过程的复杂性以及竞争产物具有相似的还原电位,实现高效且选择性的二氧化碳光催化还原制甲烷仍是一项重大挑战。
迄今为止,人们已投入大量精力将助催化剂负载于半导体载体上,并构建基于半导体的异质结构以形成双金属活性位点。这些策略有望打破传统比例关系,使各种中间体按顺序反应,最终生成甲烷。不幸的是,典型助催化剂因表面能高,在长期运行中普遍存在纳米颗粒团聚和结构不稳定的问题。同时,传统基于异质结构的光催化剂通常涉及二次组分的随机沉积,且其相对庞大的形貌给构建有利的异质界面带来极大困难,从而导致电荷转移缓慢、界面双金属位点不明确。此外,为追求高效的甲烷生成,通常会引入三乙醇胺和甲醇等牺牲剂以促进电子转移并绕过缓慢的水氧化过程,这会降低系统的可持续性并增加额外成本。因此,开发一种可行策略以优化界面电子转移并构建明确定义的串联催化中心,从而实现无需牺牲剂的选择性二氧化碳光催化还原制甲烷,显得至关重要。
将光催化二氧化碳还原制甲烷与生物质衍生醇的氧化相结合,为消除对牺牲剂的依赖并最大化利用光生电子-空穴对提供了一种有前景的策略。 然而,在当前太阳能驱动二氧化碳还原与生物质转化相结合的研究中,由于甲烷反应路径复杂且C-H键活化缓慢,选择性二氧化碳光催化还原制甲烷仍难以实现,一氧化碳成为主要产物。因此,迫切需要一种能精确调控的界面结构,以同时介导二氧化碳还原制甲烷和生物质衍生醇氧化的不同反应路径。最近,金属有机框架材料(MOFs)因其高孔隙率、优异的气体吸附能力和网状结构而闻名,已成为结合半导体纳米团簇构建异质结构的理想平台。同时,基于MOF的异质结构中的界面空位在调节异核原子间相互作用方面展现出巨大潜力,可进一步优化理想的界面电荷转移路径以构建Z型异质结构,并促进形成具有不同电荷富集的双金属位点。这种强耦合的界面结构与串联催化中心相结合,为同时实现二氧化碳光催化还原制甲烷和烷氧基C-H键氧化产生协同效应提供了巨大可能性。
本研究中,我们报告了一种精心设计的方法,即通过在富含空位的MOFs中填充半导体纳米团簇来构建非对称双金属催化中心,实现选择性二氧化碳光催化还原制甲烷并伴随苯甲醇(BA)氧化(图1)。我们选择富含氧空位的Mil-125(Ti)-NH₂/Fe₂O₃(记为Milv/Fe₂O₃)异质结构作为光催化二氧化碳还原和苯甲醇氧化的实例,因其制备相对简单且铁离子具有较高的催化倾向。选择苯甲醇是因为它可从多种天然生物质和有机资源中获取,并且也是合成精细化学品的重要中间体。具体而言,高角度环形暗场扫描透射电子显微镜(HAADF-STEM)和电子能量损失谱(EELS)映射显示,Fe₂O₃团簇明显分布在富含空位的MOFs中。原位实验和同步辐射X射线吸收光谱(XAS)表明,界面氧空位触发形成新的Fe-O键,作为快速电子转移路径以生成Z型异质结构。此外,氧空位的存在促进了Milv和Fe₂O₃之间的强界面相互作用,从而生成非对称Ti-Fe双金属位点并诱导d带中心上移。与Mil-125(Ti)-NH₂(记为Mil)和富含氧空位的Mil-125(Ti)-NH₂(记为Milv)相比,优化后的Milv/Fe₂O₃异质结构实现了高效二氧化碳光催化还原制甲烷,在450 nm波长下具有87.0%的显著选择性和2.8%的表观量子效率(AQE),同时实现了苯甲醇向苯甲醛(BZA)的近100%转化。理论计算和原位光谱表明,Ti-Fe双金属位点通过d-p轨道耦合协同稳定CO中间体,促进其进一步加氢生成CHO以合成甲烷,而Fe₂O₃产生的光生空穴则氧化苯甲醇的α-C-H键以生成苯甲醛。我们还合成了其他基于MOF的异质结构,以证实非对称双金属界面在二氧化碳光催化还原制甲烷并伴随苯甲醇氧化方面的普适性。本研究为优化界面电荷转移和构建串联催化中心以开发太阳能驱动二氧化碳还原与C-H键活化相结合的策略提供了精细设计思路。
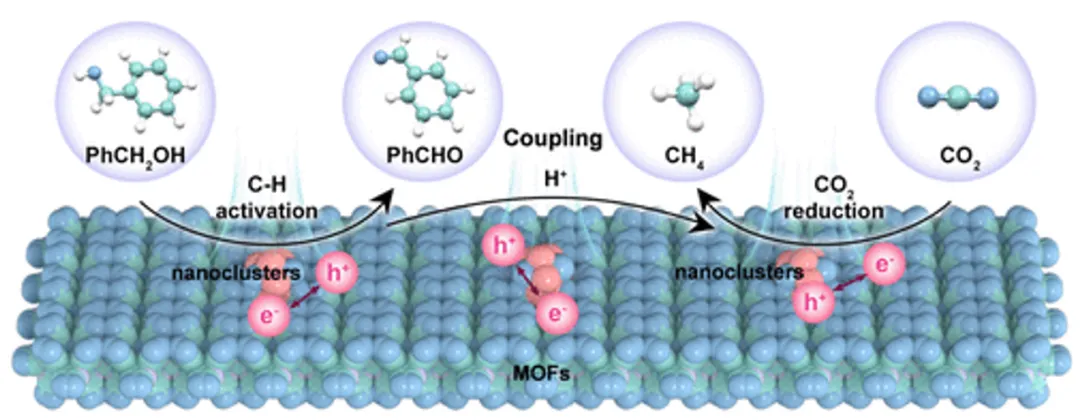
图1. 用于CO2光还原为CH4并耦合C-H键活化的非对称双金属界面的示意图:将半导体纳米团簇锚定在富含空位的MOF中构建非对称的双金属界面,可以同时实现CO2光还原为CH4并耦合C-H键活化以进行苯甲醇氧化为苯甲醛。
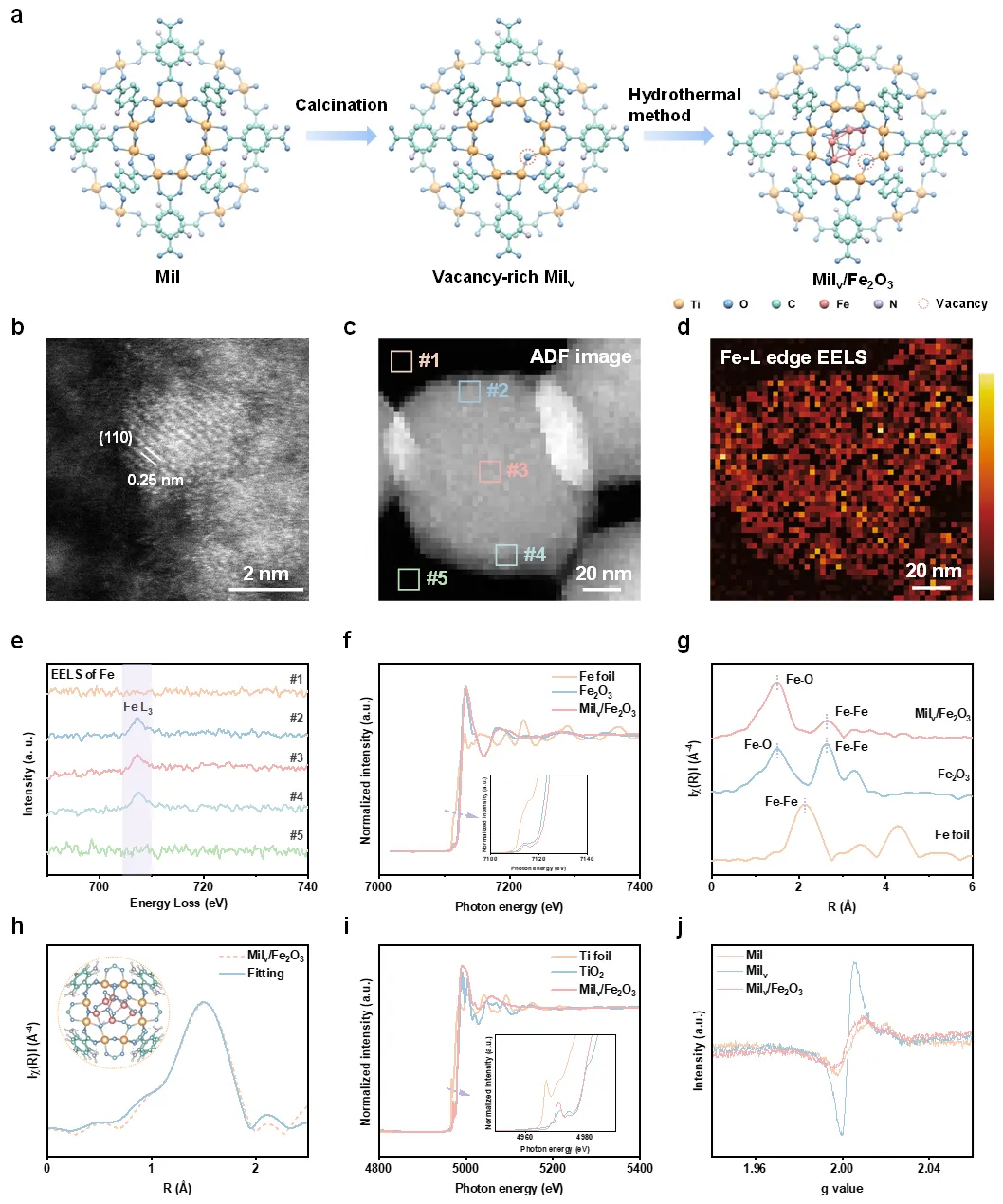
图2. MOFs/纳米团簇异质结的形貌和结构表征。通过STEM和同步辐射证明MOF担载的Fe2O3纳米团簇可以形成新的Fe-O键。
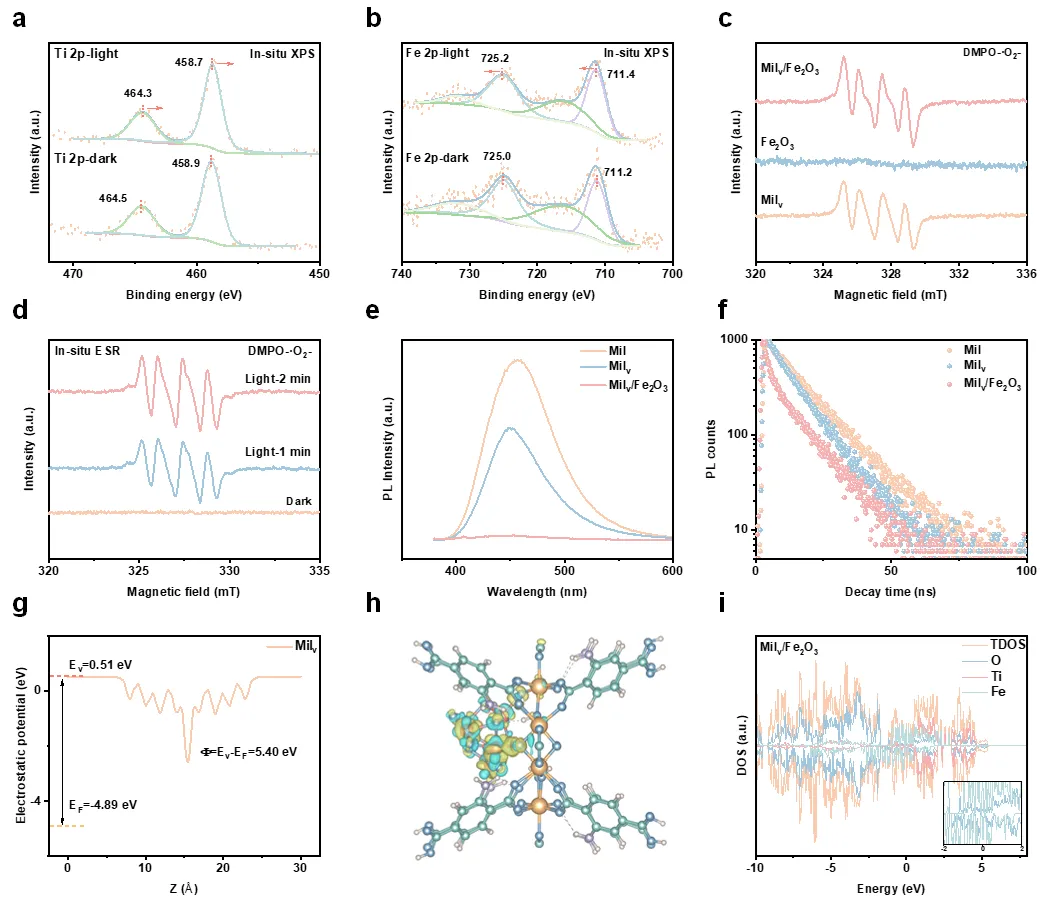
图3. 界面载流子转移的动力学研究。原位实验表征和光谱学实验证明Milv/Fe2O3异质结构可以实现载流子动力学的快速转移。
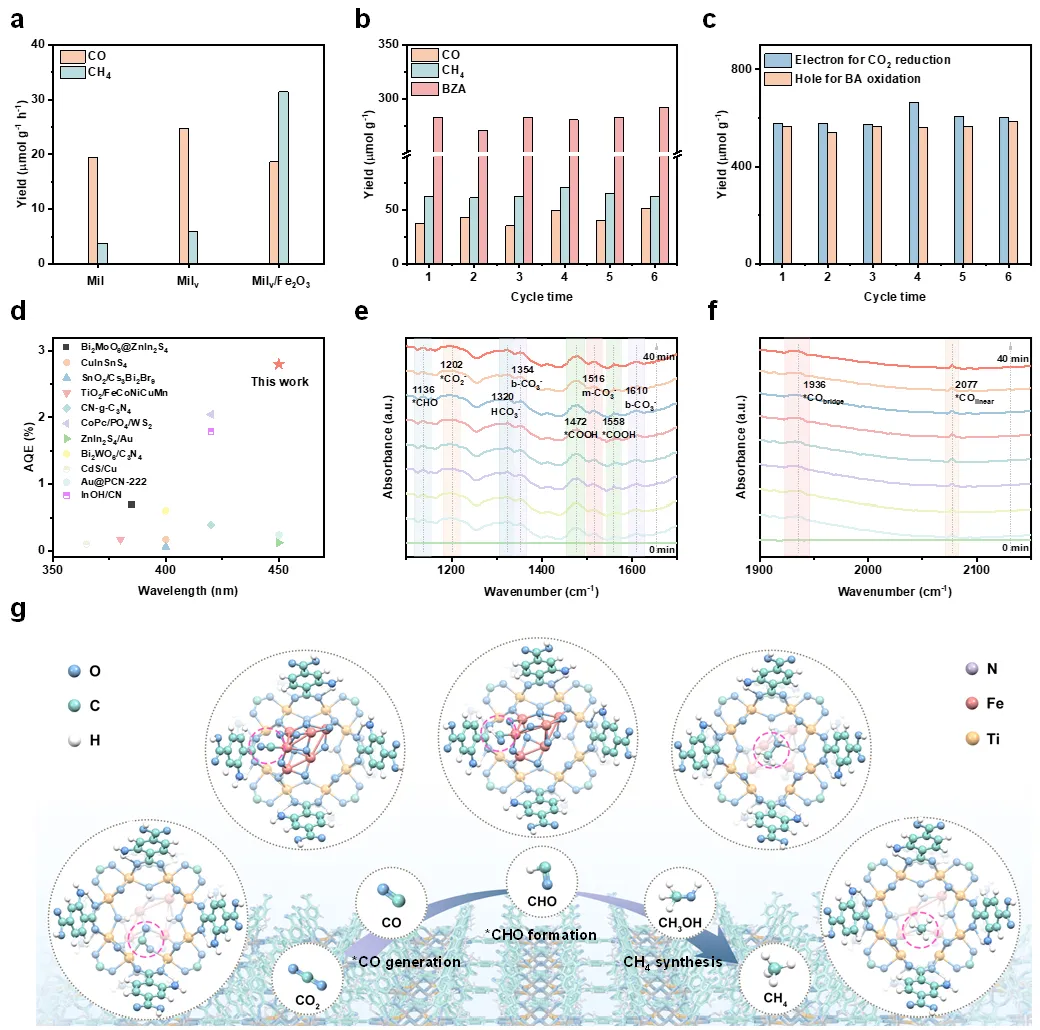
图4. 光催化CO2还原和苯甲醇C-H氧化的性能和过程研究。催化结果表明,该体系可以实现CO2还原为CH4以及耦合苯甲醇到苯甲醛的C-H氧化。
综上所述,本研究报告了一种通用策略,即通过将半导体纳米团簇锚定在富含空位的金属有机框架材料(MOFs)中,构建非对称双金属活性中心,实现高效二氧化碳光催化制甲烷与C-H键氧化偶联。原位实验与理论计算表明,界面空位可诱导形成新化学键,作为原子级精确的Z型电荷转移路径。此外,界面Ti-Fe双金属活性位点可通过d-p杂化稳定CO中间体并生成CHO中间体,最终在450 nm波长下实现2.8%的表观量子效率(AQE)及87.0%的甲烷选择性。同时,Fe₂O₃产生的光生空穴可通过C-H键活化将苯甲醇(BA)完全选择性(100%)氧化为苯甲醛(BZA)。这种用于太阳能驱动二氧化碳与生物质衍生醇串联转化的非对称界面双金属位点,凸显了光生电子与空穴同时利用的独特协同效应。本研究为通过理性设计非对称双金属界面优化电子转移路径及级联催化中心、推动先进催化发展提供了新思路。








 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
