南京师范大学付更涛/日本东北大学李昊AM | 原子铕调控乙腈吸附构型实现长寿命电合成乙胺!
- 2026-06-29 16:11:03

乙腈电催化加氢为乙胺合成提供了一条可持续的路径。
然而,要在此过程中实现高选择性,必须精细调控质子供给以抑制析氢副反应,而这与工业级电流密度下的质子需求常存在矛盾。

在本文中,作者设计并开发了一种新型高效乙腈电催化加氢催化剂——在氧化亚铜纳米针上修饰稀土铕原子,实现了安培级电流下高效、持久的乙腈电催化加氢反应。
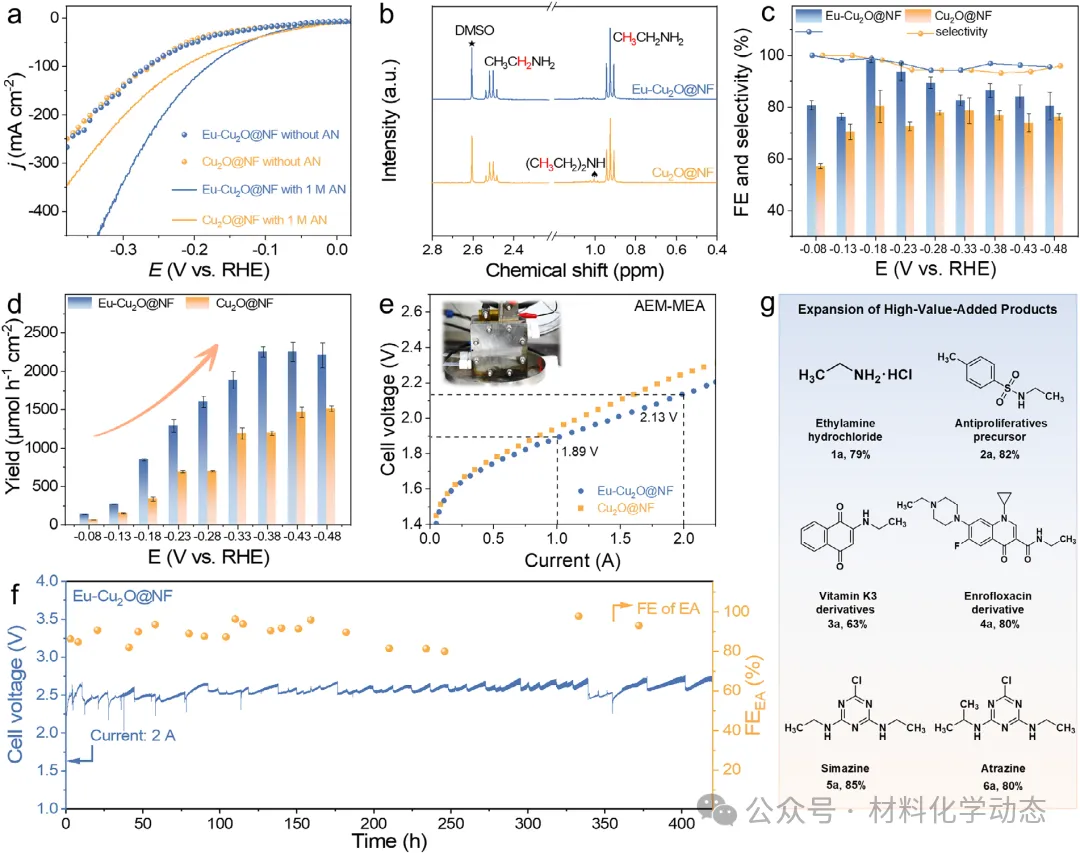
优化后的铕-氧化亚铜催化剂乙胺法拉第效率高达98.1%,产率显著提升至2253.2 µmol h-1 cm-2,优于纯氧化亚铜体系。
值得注意的是,在阴离子交换膜电解槽中,铕-氧化亚铜催化剂能在2 A电流下连续稳定运行420 h,这是目前工业电流条件下报道的最长稳定运行时间。
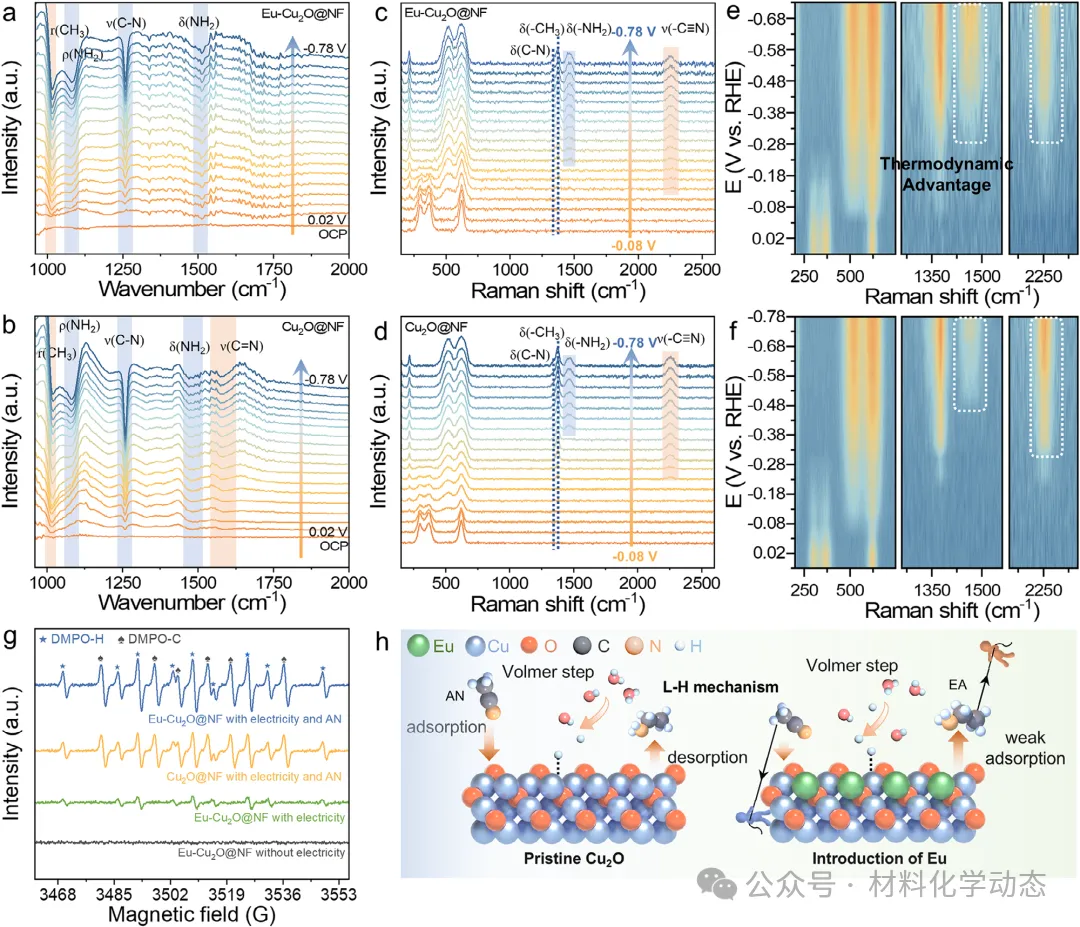
原位表征与理论计算表明,铕的引入调控了铜位点的电子结构,使乙腈吸附构型从平躺的多位点π吸附转变为垂直的氮端吸附。
这种吸附位点的重构降低了亚胺加氢步骤的能垒,导向了理想的质子加成路径,同时增强质子加成动力学以抑制析氢反应。
该研究为稀土调控乙腈加氢机制提供了基础理论依据,标志着乙胺安培级电合成技术迈出关键一步。
作为一种在工业合成中被广泛使用的基础化学品,乙胺(EA)是制药、农用化学品、染料、乳化剂和精细化学品生产中的关键组分和中间体。EA的传统合成路线,例如腈的还原或醛/酮的还原胺化,通常需要在高温高压条件下进行多相热催化,这些方法存在能耗高、操作程序复杂、依赖危险氢源(例如H2)以及产生不希望的副产物(包括二乙胺(DA)和三乙胺(TA))等问题。乙腈(AN)的电催化加氢(AN-ECH)已成为一种有前景的替代方案,该方法利用可再生电力作为驱动力,并以水或质子作为绿色氢源,能够在常温常压下实现AN向目标胺的选择性加氢。然而,要将AN-ECH技术推向工业化应用水平,需要在膜电极组件(MEAs)中以高电流密度运行,而这仍面临严峻挑战,包括严重的传质限制、析氢反应(HER)的竞争以及腈还原本身固有的缓慢动力学。应对这些挑战需要开发高效的电催化剂,使其能够在工业级电流密度下长期运行,同时保持对EA生成的高选择性。
尽管铜基催化剂已被用于AN-ECH研究,但大多数工作主要集中在调控质子吸附行为以平衡ECH与HER。然而,这类以质子为中心的策略往往由于固有的动力学和传质限制,难以在工业电流密度条件下维持高速率的腈还原。AN电催化加氢生成EA涉及多个连续的加氢步骤,其中亚胺中间体(CH3CH=NH*)的加氢是一个关键的基本步骤。这一步骤尤其具有挑战性,因为亚胺物种的吸附行为复杂,通常以平面π模式跨多个金属位点吸附。这种多位点吸附要求在加氢步骤中同时涉及两个H原子,这严重限制了可用的活性位点密度,并增加了中间体扩散和H配位相关的能量消耗。在低H覆盖度下,反应因缺乏氢供体而受阻;在高覆盖度下,HER则占据主导地位。因此,这一步骤成为动力学上的限制因素,并极易受到传质限制和竞争吸附的影响,导致在高电流下性能下降。
解决这一问题的关键策略在于调控关键亚胺中间体的吸附模式,例如从多位点π吸附转变为N端垂直吸附。这种转变将释放因水平吸附而被阻塞的相邻位点,促进吸附氢的更高效利用,并绕过缓慢的多位点H转移机制。更重要的是,通过为H沿碳加氢提供优先且快速的消耗途径,这种构型能有效降低表面H覆盖度,从而抑制H-H偶联并阻碍HER。因此,实现亚胺中间体从平面到垂直吸附构型的转变,是实现在安培级电流密度下高效稳定EA合成的核心。
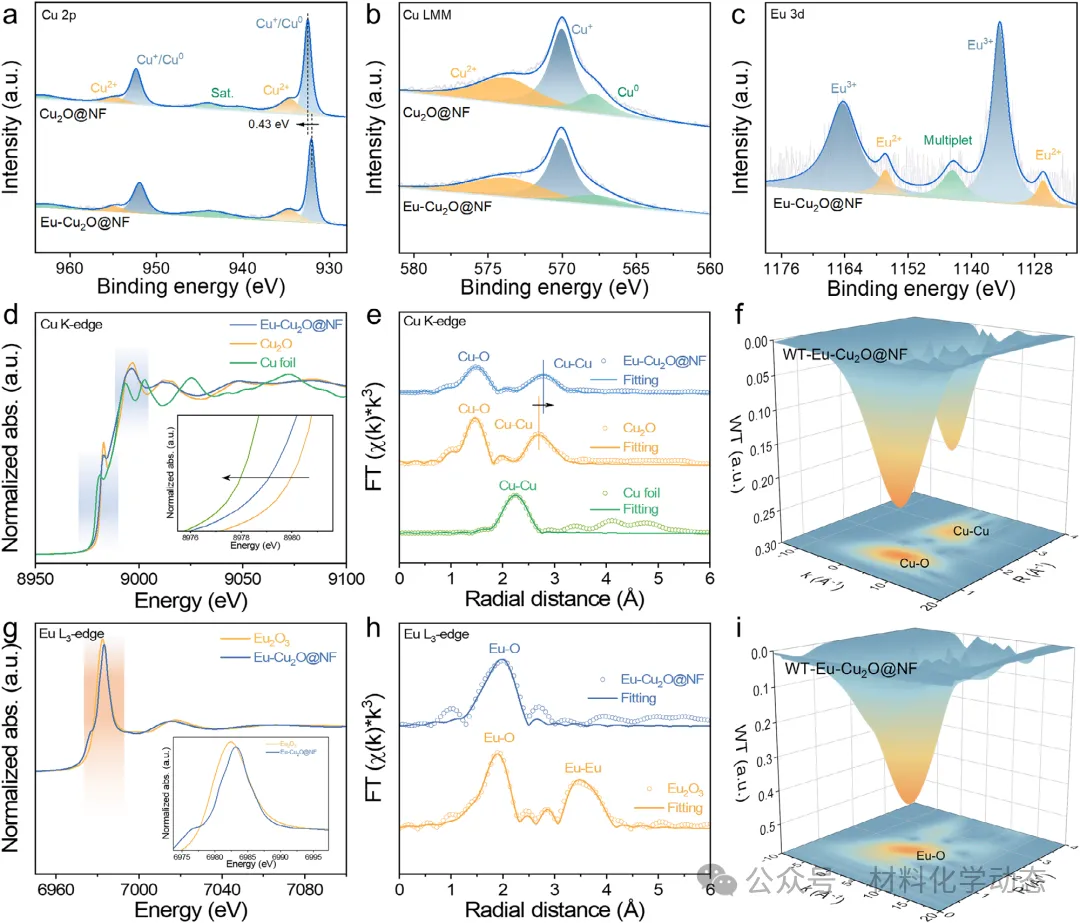
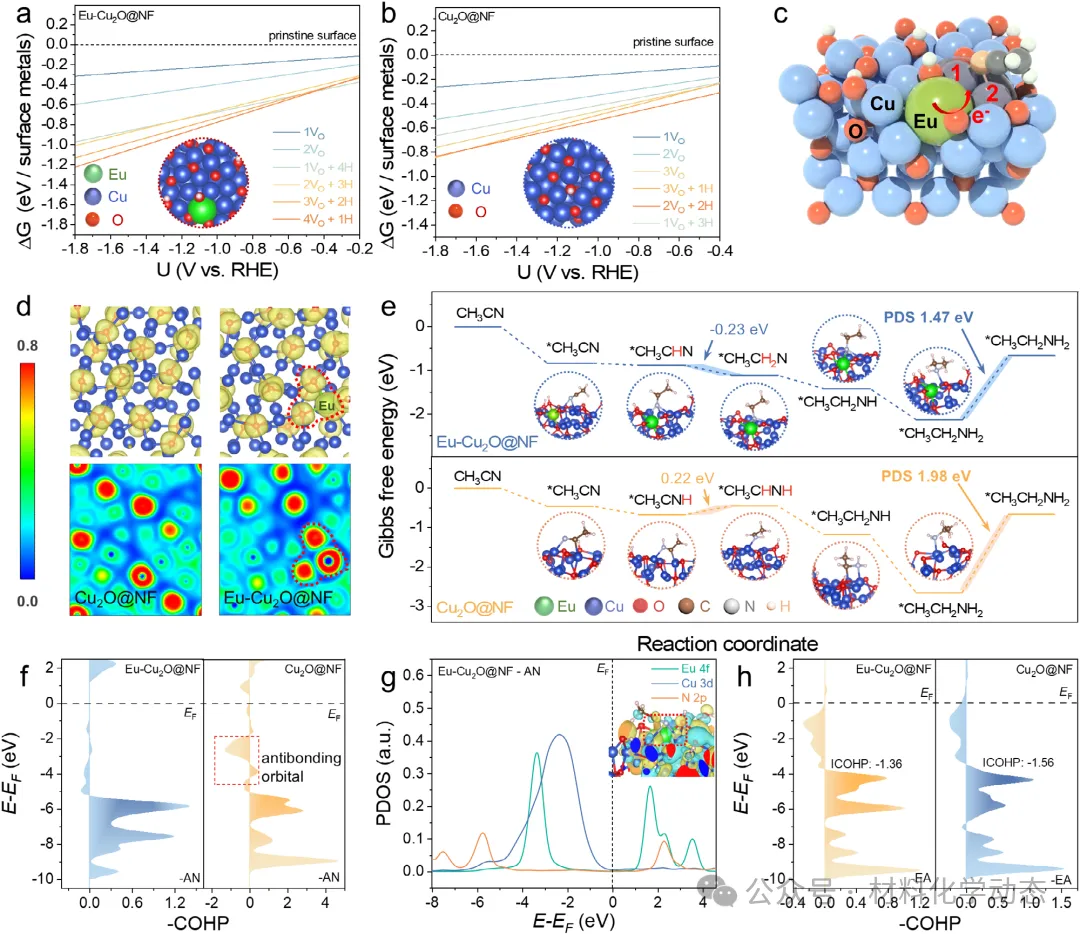
初始AN分子的吸附构型,特别是其N端垂直吸附,在后续加氢路径中起着决定性作用,因为它能诱导后续亚胺中间体形成匹配的吸附几何结构。由于从AN到亚胺存在显著的连续演化关系,AN最初的N端吸附有利于C≡N键的极化,并且更重要的是,建立了一个电子和几何微环境,引导生成的亚胺中间体也采取类似的垂直N端吸附模式。关键中间体的吸附行为本质上由催化金属位点的电子结构所决定。战略性地调控局部电子环境是增强电催化剂设计中结构-性能关系的关键途径。
稀土(RE)元素凭借其可调的4f轨道和独特的电子特性,为裁剪主体催化剂的电子结构提供了强有力的手段。Eu(1.20)与Cu(1.90)之间显著的鲍林电负性差异,通过氧桥连的键合模式诱导了明显的电子转移。这种电子重分布理论上能有效优化反应中间体的吸附能,从而实现对AN吸附构型及后续加氢路径的精确调控。然而,此类调控的通用且有效策略仍鲜有探索。
作者报道了一种负载在泡沫镍上的稀土铕(Eu)修饰的氧化亚铜纳米针电催化剂(Eu-Cu2O@NF),用于阴离子交换膜(AEM)电解槽中的乙腈电催化加氢反应。该催化剂在工业级安培电流密度下表现出卓越性能,并实现了迄今为止报道的最长运行稳定性。Eu-Cu2O@NF电极展现出优异的电催化性能,在-0.38 V下,乙胺(EA)的法拉第效率(FE)高达98.1%,产率达到2253.2 µmol h-1 cm-2,显著优于未修饰的Cu2O@NF电极在所有测试电位下的表现。当应用于AEM电解槽时,Eu-Cu2O@NF电极在2 A的工业相关电流下连续稳定运行超过420 h,过电位和EA法拉第效率几乎无衰减。
结合原位/非原位表征与密度泛函理论(DFT)计算,本文阐明了性能提升的内在机制:Eu的引入调控了Cu活性位点的电子结构,促使关键乙腈(AN)中间体的吸附构型由C-N多点π吸附转变为N端垂直吸附。这一吸附构型的改变降低了加氢步骤的能垒,诱导亚胺中间体同样采取N端垂直吸附,并引导反应沿最优质子化路径进行。由此增强的质子化动力学使得催化剂在较低过电位下即可实现高效转化,从而在高电流密度下保持高活性与长期稳定性。
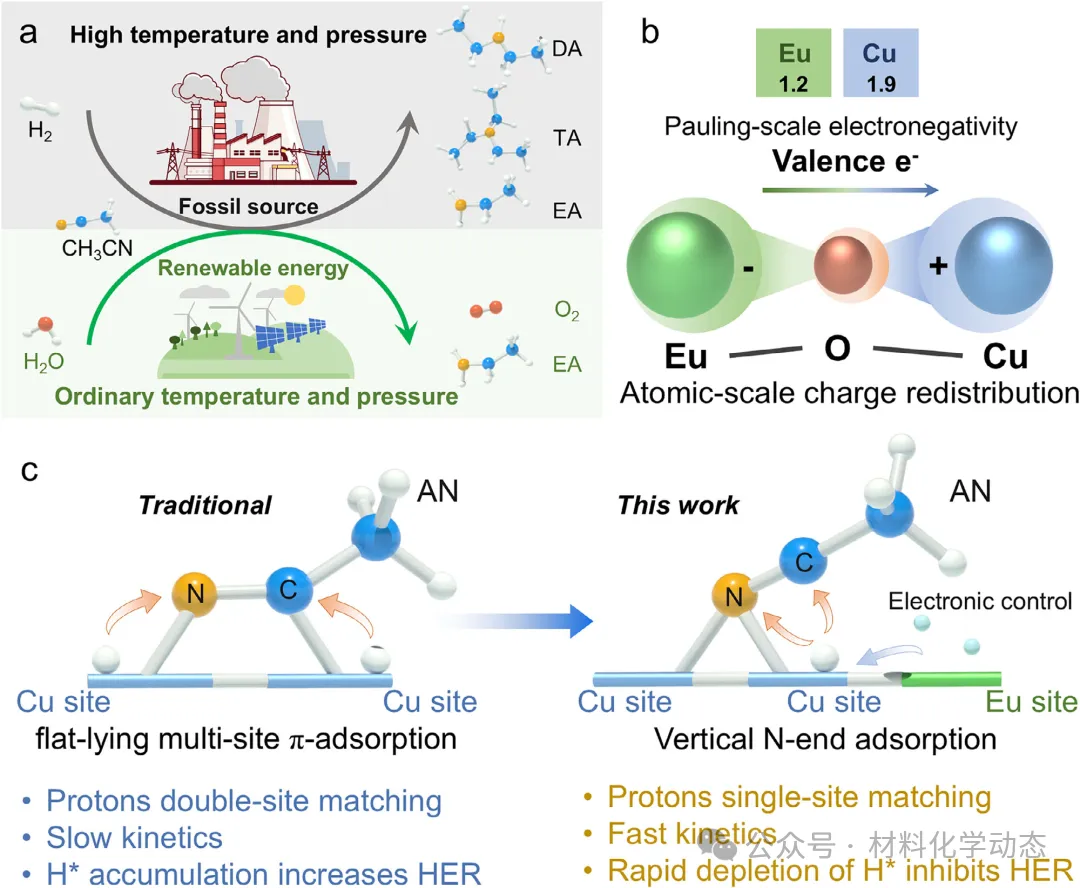
图1:(a) 传统热催化、催化与电催化AN加氢路径的比较。热催化路线常因选择性低而产生不希望的副产物,而电化学方法则能实现更绿色且高选择性的EA合成。(b) Eu掺杂诱导的金属位点电子结构调制,展示了其调控关键反应中间体吸附构型的潜力。(c) 提出的Eu介导的AN中间体从平面π吸附向垂直N端吸附转变的机制,有助于高效质子化并抑制竞争性析氢反应。
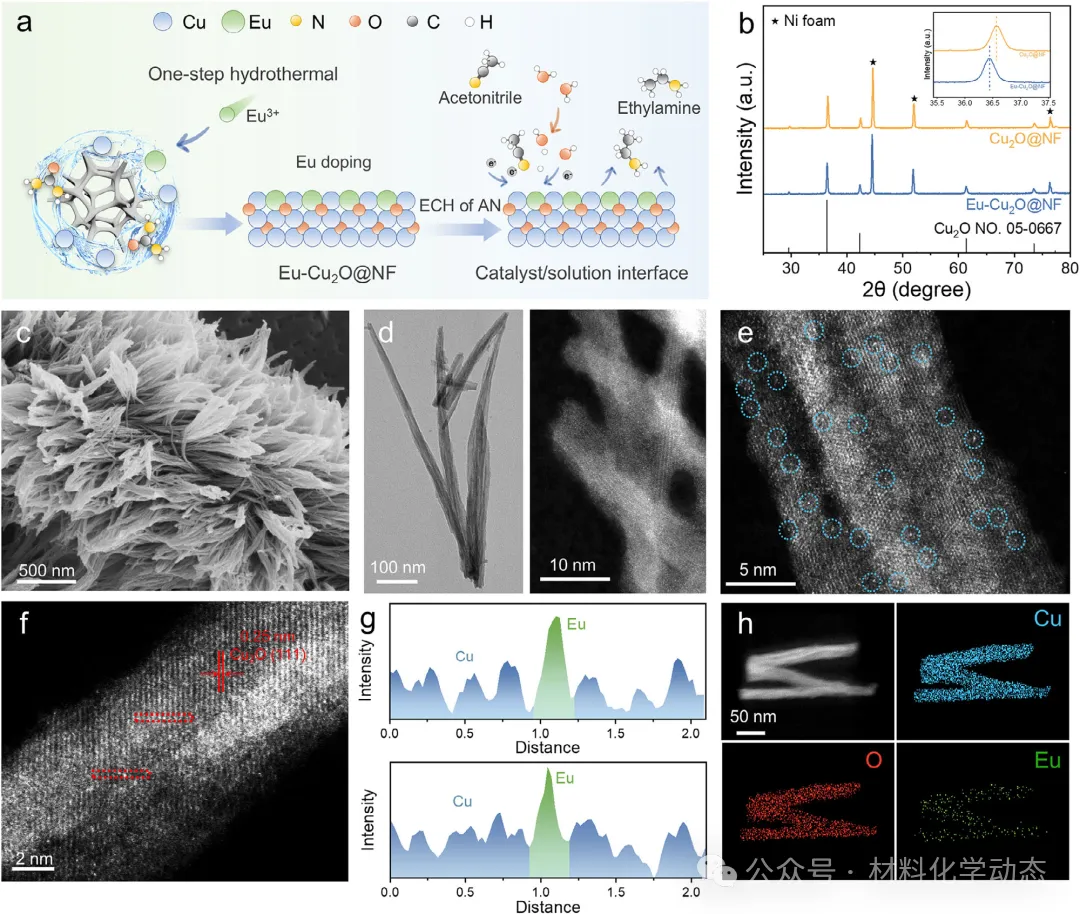
综上,作者成功开发了一种原子级铕(Eu)修饰的氧化亚铜纳米针催化剂(Eu-Cu2O@NF),用于在阴离子交换膜(AEM)电解槽中高效、稳定地电催化加氢乙腈(AN-ECH)合成乙胺(EA)。
通过将具有独特电子结构的稀土Eu原子精准掺杂到Cu2O晶格中,催化剂的电子结构得到显著调控,从而巧妙地将乙腈分子的吸附构型从平躺式的多点π吸附转变为垂直的氮端吸附。
这一关键的构型转变不仅降低了关键亚胺中间体加氢步骤的能垒,优化了质子加成路径,还加速了加氢动力学,有效抑制了竞争性的析氢反应(HER)。
得益于此,Eu-Cu2O催化剂展现出卓越的性能:在-0.38 V下,乙胺的法拉第效率高达98.1%,产率达到2253.2 µmol h-1 cm-2。
更重要的是,该催化剂在工业级电流(2 A,约0.5 A cm-2)下于AEM电解槽中连续稳定运行超过420小时,是目前报道的在安培级电流下最稳定的AN-ECH体系。
综合原位表征和理论计算,研究清晰地揭示了Eu原子作为电子供体,通过调制Cu活性位点的电子环境来实现吸附构型切换的内在机制,为理解稀土元素在电催化中的作用提供了深刻的见解。



