南京工业大学Adv. Sci:通过铁掺杂反转金属磷化物异质结中的界面电场进行大电流析氧反应
- 2026-07-06 16:43:18

1、文章亮点
1.HER催化剂1000mA/cm²下过电位266mV,OER催化剂1000mA/cm²下过电位274mV;二者组装的电解池1000mA/cm²下电压1.724V(工业条件1.620V),均稳定运行150h。
2.Fe掺杂反转并扩大金属磷化物异质结的界面电场,将富电子Ni位点转为缺电子位点,实现HER/OER活性切换。
3.原位生长无粘结剂结构,电荷转移快;法拉第效率超97%,能耗低于商用催化剂,适配工业大电流水分解场景。
2、全文速览
开发能够在高电流密度下以低过电位运行的非贵金属电催化剂的工业应用具有挑战性。多相双金属磷化物引起了人们的极大兴趣。尽管析氢反应(HER)性能很高,但普通的析氧反应(OER)性能阻碍了它们的实际应用。结果表明,Fe掺杂反转并增大了异质结的界面电场,使HER的H变为O中间OER有利结合位。具体地说,泡沫镍自担载异质结催化剂(CoP@Ni2P/Nf和Fe-CoP@Fe-Ni2P/Nf)的合成比较容易。它们只需要266和274 mV的过电位就可以分别为HER和OER驱动1000 mA cm−2(J1000)的大电流密度。此外,配备这些电极的分水池只需要1.724V的电压就可以驱动J1000,具有良好的耐用性,显示了工业应用的潜力。这项工作为异质结催化剂的界面工程提供了新的见解。
3、背景介绍
电化学分解水所产生的氢能受到贵金属基电催化剂(如用于HER的铂和用于OER的IrO2/RuO2)的依赖以及阳极析氧反应(OER)的缓慢动力学的限制。尽管人们已经做出了巨大的努力来开发各种非贵金属催化剂,但用这些催化剂构建的裂水电池往往存在驱动电压高或在大电流密度下稳定性差的问题,不能满足工业应用的要求。为了在给定的电压下通过增大电活性表面积来提高电流密度,合成的电催化剂被沉积在3D电极上,如Ni,Co和Fe泡沫。在这些自支撑电极上原位生长电催化剂不需要粘结剂,简化了合成,提高了电荷转移和稳定性。
过渡金属磷化物(TMP)具有比金属氧化物更好的导电性、较高的HER催化活性和较低的成本,因而引起了人们的极大关注。具有异质结的双金属磷化物表现出更好的性能,因为建立的界面电场促进了电子的迁移,导致了带负电荷的活性中心有利于氢中间体的吸附。另一方面,由于带负电荷的活性中心不利,普通金属磷化物的OER性能进一步恶化为了解决这个问题,研究人员将双金属磷化物异质结中的一个相替换为具有比金属磷化物低功函数(Φ)的OER活性相(例如,金属氧化物、金属(氧)氢氧化物),以产生OER/HER双功能催化剂。但制造过程复杂(因此成本较高)。此外,TMP与氧化物或(氧)氢氧化物的偶联会导致金属位上的电子离域,阻碍异质结界面的电子转移。认为,通过建立适当的界面电场,扩大两相之间的功函数差,创建缺电子的活性中心,简单地通过杂原子掺杂来促进电子转移,可以改善TMP异质结催化剂的OER性能。
在这里,在泡沫镍上生长了过渡金属磷化物异质结(CoP@Ni2P),它只需要266 mV的小过电位就可以为她驱动-1000 mA cm−2(j-1000)的大电流密度。将Fe掺杂到CoP@Ni2P中,构建了一种只需274 mV的小过电势即可驱动J1000的OER电极(Fe-CoP@FeNi2P/Nf)。实验和密度泛函理论(DFT)结果表明,Fe的掺杂反转并放大了界面电场,使CoP@Ni2P/Nf中的富电子Ni位转变为贫电子位,有利于氧中间体的吸附。配备这些自支撑电极的整体裂水电池可以在1.724 V的低电压下驱动J1000,并具有出色的长期稳定性,提出了一种新的异质结催化剂界面工程策略,即通过掺杂金属元素来调节界面电场。
4、图文解析
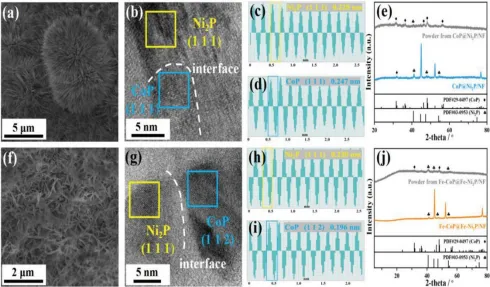
图1.a-e)CoP@Ni2P/Nf和f-j)Fe-CoP@Fe-Ni2P/Nf的表征A,f)扫描图像。B,g)HRTEM图像(虚线表示Ni2P和CoP之间的界面)。(c和d;h和i)Ni2P面和CoP面的快速傅立叶逆变换(FFT)的行扫描。E,j)X射线衍射谱。
对于CoP@Ni2P/Nf,针状CoP均匀地分布在Nf表面(图1a)。CoP@Ni2P/Nf截面的EDS图谱显示P掺杂到Nf骨架中,形成了一层薄薄的Ni2P/Nf。Co和P在Ni2P/Nf上的均匀分布表明磷化钴和磷化镍之间形成了异质结。高分辨率电子显微镜图像进一步显示了两种金属磷化物之间的界面,并显示了分别对应于Ni2P和CoP的(111)面的0.220 nm和0.247 nm的晶格条纹间距(图1b-d)。CoP@Ni2P/Nf的X射线衍射图(图1e)显示出三个Ni2P的特征峰,类似于Ni2P/Nf。超声处理CoP@Ni2P/NF得到的粉末在31.5、36.3、46.2、48.1和56.2°处出现峰值,分别对应于CoP的(0 1 1)、(1 1 1)、(1 1 2)、(2 1 1)和(0 2 0)晶面。这些结果证实了CoP@Ni2P/Nf的异质结构。
与针状CoP不同,Fe-CoP@Fe-Ni2P异质结是片状的,这是由于高价Fe3+的加入改变了晶体结构(图1f)。如Fe-CoP@Fe-Ni2P/Nf截面的EDS图所示,Co、Ni、Fe、P均匀地分散在Nf上。此外,经超声波处理的Fe-CoP@FeNi2P粉末的透射电子显微镜图谱也显示,Fe在CoP和Ni2P相中均匀分布,表明Fe是相同的同时掺杂到CoP和Ni2P相中。Fe-CoP@Fe-Ni2P/Nf的HRTEM图像显示了CoP和Ni2P之间的界面,晶格条纹分别对应于Ni2P和CoP的(1 1 1)和(1 1 2)面(图1g)。没有证据表明Fe基磷化物的存在(图1h,I),证实了Fe-CoP@Fe-Ni2P/Nf中Fe作为掺杂剂的存在。超声法制备的FeCoP@Fe-Ni2P/Nf和自支撑Fe-CoP@Fe-Ni2P/Nf粉末的X射线衍射谱仅显示CoP和Ni2P峰(图1J),这与HRTEM观察到的结果一致。
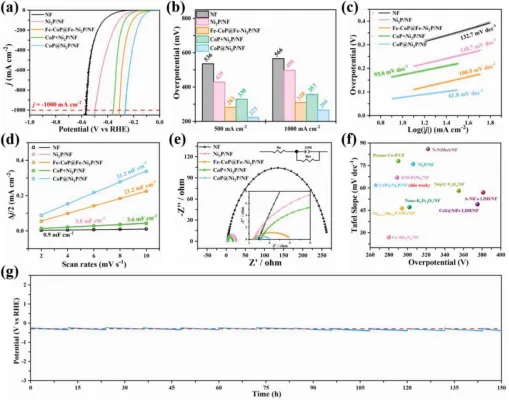
图2 HER的:a)极化曲线。B)j-500和j-1000处的过电位。C)塔菲尔地块。D)双层电容(CDL)。E)EIS曲线(插图显示等效电路)。F)在J-1000时与其他自负载型电催化剂的Tafel斜率和过电位的比较)在J-1000时CoP@Ni2P/Nf的计时电势曲线(定期更换电解液,每隔12h更换一次)。
在室温下,三电极系统在1M的KOH溶液中评价了样品的HER性能。如图2a所示,CoP@Ni2P/Nf表现出最高的HER性能,低过电位为266 mV,达到j-1000,远低于纯Nf、Ni2P/Nf和Fe-CoP@FeNi2P/Nf(图2b)。此外,CoP@Ni2P/NF表现出最低的塔菲尔斜率为61.8mVdec−1(图2c),这表明HER的动力学和Volmer-Heyrovsky电催化过程很快。CoP@Ni2P/Nf显示最高的CDL值为31.2mF cm−2(图2d),表示最大的活性区域。此外,电荷转移电阻(RCT)是通过拟合电化学阻抗谱(EIS)数据确定的(图2E)。CoP@Ni2P/Nf显示出最低的RCT值(1.5Ω),表明电荷转移速率很快。相比之下,CoP和Ni2P/Nf的机械混合物在j-1000时具有较高的过电位(357 MV),较高的塔菲尔斜率(93.6mV dec−1),较低的Cdl(3.6mF cm−2),以及RCT值(14.6Ω)高于CoP@Ni2P/Nf,表明原位生长的异质结构是催化剂具有优异催化活性的关键。总而言之,CoP@Ni2P/NF优异的HER性能归功于其高的本征催化活性、快速的OER动力学和低的电荷转移阻力,这一点优于先前报道的自负载型电催化剂(图2f)。为了评价CoP@Ni2P/NF的长期稳定性,在J-1000下进行了计时电位法测量。如图2g所示,经过150小时的测试,CoP@Ni2P/NF的过电位没有显著增加。虽然长期试验后催化剂表面变得粗糙,但指定为金属磷化物的X射线衍射图中的特征峰没有移动,拉曼光谱中也没有出现新的峰,表明在HER过程中没有产生新的物种。CoP@Ni2P/Nf粉末的X-射线衍射谱中的另一个小峰被指定为Co(OH)2。研究表明,CoP在KOH电解液中可以被轻微氧化成Her失活的Co(OH)2。CoP@Ni2P/NF在高电流密度下表现出良好的长期稳定性,因此是一种适合于工业分水应用的阴极。
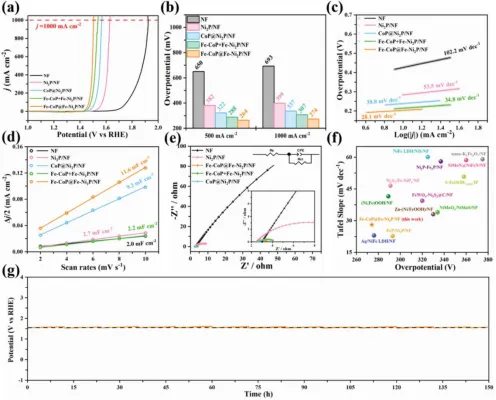
图3.样本的OER活动A)极化曲线。B)j500和j1000处的过电位。C)塔菲尔斜率。D)双层电容(CDL)。E)EIS曲线(插图显示等效电路)。F)与其他自负载型电催化剂在J1000.g下的Tafel斜率和过电位的比较)在J1000下150h(定期更换电解液,每隔12小时更换一次电解液)的计时电势曲线。
Fe-CoP@Fe-Ni2P/Nf在J1000下表现出比CoP@Ni2P/Nf(337 MV)更高的OER活性(274 MV)(图3b)。值得注意的是,进一步增加了Fe/Co的摩尔比在合成过程中,铁掺杂异质结(Fe-CoP@Fe-Ni2P/Nf)的过电势略有增加,这可能是因为过量掺杂到金属磷化物中对电子转移产生不利影响,并且Nf被Fe离子过腐蚀。此外,Fe-CoP@Fe-Ni2P/Nf的−(11.6mFcm−2)(图3D);和更低的RCT值(1.1Ω)(图3E)比其他样品具有更高的CDL(11.6mFcm RCT2)(图3d)值得注意的是,Fe-CoP和Fe-Ni2P/Nf的机械混合物比Fe-CoP@Fe-Ni2P/Nf有更高的过电位(307 MV),更高的塔菲尔斜率(34.8 mVdec−1),更低的CDL(2.2mF cm−2)和更高的RCT值(1.6Ω),证明了原位生长异质结的重要性。总而言之,Fe-CoP@Fe-Ni2P/NF优异的OER性能归功于其高的本征催化活性、快速的OER动力学和低的电荷转移阻力,这超过了以前报道的自负载型电催化剂(图3f)。如图3G,与最近报道的电催化剂相比,Fe-CoP@Fe-Ni2P/NF在150h计时电位法后的过电位仅增加了20 mV,表现出良好的长期稳定性。经过长期测试,催化剂表面只变得稍微粗糙一些。由于金属(氧)氢氧化物的标准热焓(ΔHf0)和吉布斯能(ΔGf0)比金属磷化物的更负,Fe-CoP@Fe-Ni2P在OER条件下应该被氧化。事实上,经过长期的OER,金属磷化物的特征X射线衍射峰几乎消失,在400到600 cm−1之间出现了一个新的可归因于Co/Ni(氧)氢氧化物的宽拉曼峰。在重建过程中,Fe仍然以掺杂的形式存在。此外,在OER试验后,从XPS测量光谱Oe-CoP@Fe-Ni2P/Nf中没有观察到与P元素对应的峰,表明P在OER过程中被浸出到溶液中。经过长期OER测试后的Fe-CoP@Fe-Ni2P/NF的HRTEM图像显示出Fe-CoOOH和Fe-NiOOH之间清晰的界面。0.239 nm的晶格条纹间距对应于NiOOH的(01 1)面(参见PDF#00-027-0956),而0.243、0.230、0.235 nm的晶格条纹间距对应于CoOOH的(0 21)、(11 1)、(0 40)面(参见PDF#00-026-0480)。此外对于Fe-CoP@Fe-Ni2P/Nf催化剂,当第一个循环中的电位从1增加到1.8V(Vvs RHE)时,电流密度总是高于随后几个循环的电流密度,这可能是因为金属磷化物催化剂向金属氢氧化物的不可逆转变所致。以前的研究中也观察到了类似的现象。在高电位下,金属氢氧化物将进一步可逆地转变为金属(氧)氢氧化物。为了验证这一点,我们在低扫描速率下进行了10个循环的循环伏安测试后,进一步测量了Fe-Ni2P的循环伏安曲线(图S12b,支持信息)。当电位从1增加到1.8V(Vvs RHE)时,由于Co2+和Ni2+被氧化成Co3+和Ni3+,在RHE的1.3-1.5V附近观察到明显的氧化峰。当电极电位从1.8V降至1V(V Vs RHE)完成循环时,在低电位(1.1~1.3V vs RHE)下可观察到一个还原峰,表明Co3+和Ni3+被还原为Co2+和Ni2+。这种可逆转变与以前的报道一致。考虑到OER是在高电位下进行的,实际的活性相是金属(氧)氢氧化物(Fe-CoOOH@Fe-NiOOH)。在这种结构重建过程中,可能会原位产生不饱和的金属中心,阳离子或阴离子物种的丢失将导致低结晶度甚至无定形相的形成,从而增加活性中心的比表面积和暴露。这可以提高OER性能。这些结果表明,Fe-CoP@Fe-Ni2P/Nf(实际上是Fe-CoOOH@Fe-NiOOH/Nf)在高电流密度下具有良好的长期稳定性,因此是一种适合于工业分水应用的阳极。
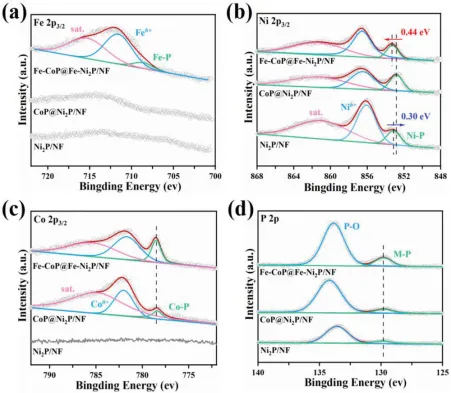
图4.Ni2P/Nf、CoP@Ni2P/Nf和Fe-CoP@Fe-Ni2P/Nf的XPS谱A)Fe 2p3/2.b)Ni2p3/2.c)Co2p3/2 d)P2p。
通过表征和密度泛函理论(DFT)计算,研究了CoP@Ni2P/NF和Fe-CoP@FeNi2P/NF对HER和OER的催化机理。X射线光电子能谱(XPS)(图4a)表明,Fe已成功掺杂到金属磷化物异质结中,Fe-CoP@Fe-Ni2P/Nf中Fe-─P键在708.6 eV处的结合能(BE)表明,Fe掺杂成功。对于Ni2P/Nf,853.12 eV处的峰来自Ni-P键,而856.08 eV处的Be对应于Ni表面氧化(图4b)。对于具有异质结构的CoP@Ni2P/Nf,Ni-P的BE下移到852.82 eV,表示负电荷增加Ni-─-P键中的OfNi是由于界面电子转移引起的。与之相比,Ni的─Pin Fe-CoP@Fe-Ni2P/Nf的Be上移至853.26 eV,表明Fe掺杂使Ni失去了电子。对于Fe-CoP@Fe-─P和CoP@Ni2P/Nf,在778.38 eV的Be来自Co─P键中的Co(图4c),而在129.85 eV的Be主要由P在金属-P键中所归属(图4d)。Fe掺杂后,两个峰没有明显的移动,表明Fe的掺杂主要影响了Ni─P中Ni位附近的电荷密度。
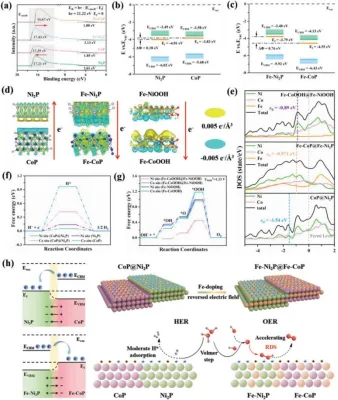
图5.(A)Ni2P、CoP、Fe-Ni2P和Fe-CoP的UPS谱。B)Ni2P和CoP的能带图(Evac:真空度。ECBM:导带最小。EF:费米能级。EVBM:价带最大值)。C)Fe-Ni2P和Fe-CoP的能带图。D)异质结(CoP@Ni2P、Fe-CoP@Fe-Ni2P和Fe-CoOOH@Fe-NiOOH)界面的电荷密度差异。E)态密度(DOS)。F)Ni2P、CoP和CoP@Ni2P的自由能图。G)OER的Fe-NiOOH、Fe-CoOOH和Fe-NiOOH@Fe-CoOOH的自由能图。H)拟议机制的图解。
当具有不同功函数的两个组分(Φ,定义为位于费米能级的电子移动到真空能级所需的能量)聚集在一起形成异质结时,将在界面上建立一个电场,从而导致电子从低Φ侧转移到高侧。基于紫外光电子能谱(图5a)反键态是由于金属原子的d轨道与H原子的1s轨道(HER)或O原子的2p轨道(OER)之间的耦合而产生的。金属位置的Ed越接近费米能级,处于反键状态的电子越少,因此中间体在金属位置上的吸附越强。CoP@Ni2P的禁带宽度比纯CoP宽,但比纯Ni2P窄。结果表明,氢中间体的吸附强度顺序为:CoP>CoP@Ni2P>Ni2P。Sabatier原理假设中间体在活性中心上的吸附既不应该太强也不应该太弱,解释了CoP@Ni2P的优越性能。同样,与纯Fe-CoP和Fe-Ni2P相比,具有中等带隙的Fe-CoP@Fe-Ni2P具有最好的OER性能。
结合UPS和UV-Vis的结果,绘制了CoP,Ni2P(图5b)和Fe-CoP,Fe-Ni2P(图5c)的能带图。CoP@Ni2P是一种典型的I型异质结,不利于电子和空穴的分离。CoP和Ni2P之间的ΔΦ为0.18 eV,为电子从CoP到Ni2P的转移提供了较弱的驱动力。总而言之,CoP@Ni2P异质结结构不利于形成不同的亲核和亲电中心。相反,Fe掺杂将I型异质结(CoP@Ni2P)转变为II型异质结(Fe-CoP@Fe-Ni2P),有利于电子和空穴的分离。此外,Fe-CoP和Fe-Ni2P之间的ΔΦ更大(0.76 eV),这表明Fe掺杂不仅改变了界面电场的方向,而且增加了非均相磷化物之间的能级差,从而有效地加速了非均相磷化物之间的能级差异Fe-Ni2P向Fe-CoP的电子转移导致了OER的亲电Ni位。
然后,构建了以CoP@Ni2P异质结为HER阴极,Fe-CoP@Fe-Ni2P/Nf,FeCoOOH@Fe-NiOOH异质结(Fe-CoP@Fe-Ni2P/Nf进行原位氧化,为OER阴极的密度泛函模型(图5d)。对于Fe掺杂的异质结,考虑到OER通常发生在催化剂表面,而XPS是一种表面分析技术,由于XPS测得Co:Fe:Ni的原子比为4.53:5.46:4.73,并且Fe均匀地掺杂到CoP和Ni2P(或CoOOH和NiOOH)中,假设Co:Fe:Ni=1:1:1。电荷密度差分析(图5d)显示了从CoP到Ni2P的明显的电子转移,导致电子在Ni位上的浓缩,这有利于H~*的吸附。Fe掺杂后,Fe-CoP@Fe-Ni2P上发生了反向电子转移,导致Ni位附近的电子亏损。在OER过程中,Fe-CoP@Fe-Ni2P被氧化为Fe-CoOOH@Fe-NiOOH。然而,界面电场的方向保持不变,表明表面重构不会对界面上的电子传递产生不利影响。如图5E所示,计算的态密度表明,与CoP@Ni2P相比,Fe-CoP@Fe-Ni2P的d向费米能级方向上移,从而有利于O中间体(*OH)的吸附。此外,FeCoOOH@Fe-NiOH态密度进一步上移,表明表面重构增强了中间体(*OH)的吸附。因此,对于OER来说,Fe-CoOOH@Fe-NiOOH是可取的,而CoP@Ni2P则适合于her。
对于CoP@Ni2P异质结中的Ni位,ΔGH*为0.048 eV(图5f),这比异质结中的Co和P位、纯CoP中的Co和P位以及纯Ni2P中的Ni和P位的ΔGH*更接近0 eV.在Fe掺杂后,CoP中的Co位和Ni2P中的Ni位都经历了ΔGH*的增加,这表明Fe掺杂破坏了HER的动力学。Fe-CoOOH@Fe-NiOOH中的界面电场与CoP@Ni2P中的界面电场相反,使得Ni位是缺电子的,因此是OER过程中吸附O中间体(*OH、*O和*OOH)的亲电中心。在FeCoOOH@Fe-NiOOH,Fe-CoOOH,Fe-NiOOH,CoOOH,NiOOH的所有金属中心中,Ni中心在氧化还原过程中的每个反应步骤中的ΔG最低,尤其是RDS(O*到OOH*的转化),这表明以Ni为活性中心,Fe-CoOOH@Fe-NiOOH的OER动力学最快(图5g)。总而言之,图5h说明了水分解的催化机理。
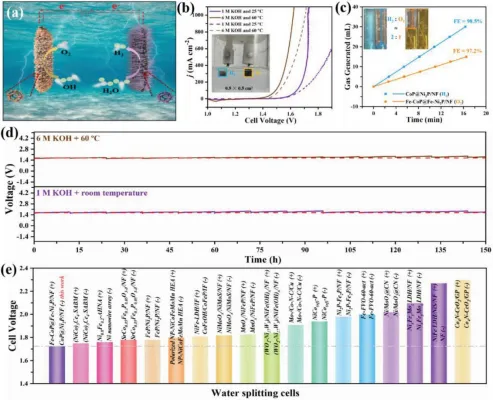
图6.样品的整体分解水的电催化活性:a)方案OFF-COP@Fe-Ni2P/Nf(+)11 COP@Ni2P/Nf(-)分水电池。B)电池在25°C的1M KOH和60°C的6M KOH中的反向极化曲线(实线:Fe-CoP@Fe-Ni2P/Nf(+)ll CoP@Ni2P/Nf(-),虚线:RuO2(+)ll PtC(-))。C)随着时间的推移产生H2(蓝线)和O2(橙线)。插图显示,生成的H2和O2分别为30和15毫升。D)在恒定j1000下,在1M KOH,25°C和6M KOH,60°C(定期更换电解液,间隔12小时)中进行150小时的计时电位法测量。E)在1M KOH中将电池电压与报道的电催化剂进行比较。
构建了一个以CoP@Ni2P/Nf为阴极,Fe-CoP@Fe-Ni2P/Nf为阳极的裂水池(图6a)。这在实验条件下(1MKOH和室温),电池只需要1.724 V的电压就可以驱动(图6b),在工业分水条件下(6 MKOH和60°C),它可以进一步降低到1.620 V。该电池在两种条件下的性能均优于商品化的RuO2(+)11PtC(-)电池,显示了其实际应用的潜力。根据测量的氢气和氧气产量计算的法拉第效率(图6c)分别为98.5%和97.2%,摩尔比接近2:1。此外,在实验和工业条件下,电池在J1000(图6d)下运行150小时后保持稳定。在实验和工业条件下,电池在性能和稳定性方面都与配备自支撑电极的最先进的水分离电池相当或超过它们(图6e)。在工业条件下(无IR补偿),电池电压OFE-COP@Fe-Ni2P/Nf(+)ll CoP@Ni2P/Nf(-)在工业条件下为1.68V,制氢的FE为98.5%,表明能耗为≈4.1kWh m−3 H2,低于商业Raney Ni电极配置的电池(2.05V达到400 mA cm−2,相应的能耗为4.9kWh m−3 H2)和美国能源部设定的目标。
5、总结与展望
综上所述,自支撑HER正极(CoP@Ni2P/Nf)和OER负极(Fe-CoP@Fe-Ni2P/Nf)都很容易合成。综合的实验和理论研究表明:1)在CoP@Ni2P异质结之间建立了界面电场,这是因为CoP@Ni2P异质结中两种磷化物(∆Φ)的功函数,使得富电子的Ni位有利于H~*在HER中的吸附;2)Fe掺杂的界面工程逆转和增大了界面电场,从而使Ni位变为缺电子位,有利于O中间体在OER过程中的吸附。配备这两个电极的整体裂水电池只需要1.724 V的低电压就可以长期稳定地驱动J1000,显示出工业应用的潜力。研究表明,通过掺杂金属元素来调节异质结催化剂的界面电场,从而改变其催化性能是一种有效的策略。
6、文章链接
《Reversing the Interfacial Electric Field in Metal Phosphide Heterojunction by Fe-Doping for Large-Current Oxygen Evolution Reaction》
https://doi.org/10.1002/advs.202308477


如需转载或投稿请联系我们:energy_catalysis@163.com
随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- 金浪南京锂电展 | 河南小威环境科技——锂电池、废旧锂电池回收线
- 南京工业大学,中国林科院林产化学工业研究所 | 周栋,尹浩然,刘定华,等:竹基活性炭强化二乙醇胺水溶液吸收CO₂
- 南京师范大学工会 2026 年教职工俱乐部(协会)会员招募开始啦!
- 南京专家来盱眙县中医院坐诊信息(3月23日-3月28日)
- 南京女孩来这里从一套搭配开始
- 南京暴走半日,
- 南京访古-南京市博物馆(朝天宫)
- 「南京非急救医疗转运指南」:医疗转运不慌张!院后转运 /康复 / 转院 / 返乡,救护车转运专业团队为您护航每一步
- 知名女演员,来南京斩鸭子连吃带打包!
- 江苏教师招聘,南京城市职业学院(南京开放大学)招聘
