2026年3月,南京医科大学团队在期刊《Journal of Clinical Investigation》(IF:13.6)在线发表题为:SHMT2 deficiency disrupts transcriptional regulation through homocysteine-mediated suppression of histone lactylation in Huntington's disease models 的高水平研究论文。
亨廷顿病(HD)是一种致命的神经退行性疾病,其特征为进行性运动功能障碍、认知下降和纹状体神经元退化,主要影响中等棘状神经元(MSNs)。尽管进行了广泛的研究,但HD发病机制中潜在的代谢脆弱性仍然知之甚少。
在本研究中,我们采用RNA测序(RNA-seq)和代谢组学分析,发现HD中一碳代谢显著失调。我们验证了线粒体一碳(mt-1C)通路中的关键线粒体酶SHMT2在HD患者来源的iPSC分化人纹状体类器官(hSOs)和YAC128小鼠中显著下调。
在功能上,药理学抑制或基因删除SHMT2会加剧突变亨廷顿蛋白(mHTT)聚集,诱导hSOs中MSN退化,并损害野生型小鼠的运动功能。相反,SHMT2过表达可以减轻HD-hSOs中的MSN退化,并改善YAC128小鼠的运动表现。在机制上,SHMT2缺乏导致高半胱氨酸(HCY)积累,HCY与AARS1相互作用并抑制组蛋白乳酸化,从而扰乱转录调控,并与神经退行性表型相关。
最后,我们证明HD临床药物氟哌啶醇能够调节SHMT2表达并恢复组蛋白乳酸化,为在HD模型中探究SHMT2依赖的代谢和表观遗传调控提供了药理学工具。这些发现强调了代谢-表观遗传轴作为HD有前景的治疗靶点。
1. 首次发现线粒体一碳代谢酶SHMT2在HD中显著下调,导致同型半胱氨酸(HCY)蓄积;
2. 阐明HCY通过竞争性结合AARS1抑制组蛋白乳酸化的新途径;
3. 证实SHMT2缺失通过HCY-AARS1-组蛋白乳酸化轴驱动纹状体神经元退变;
4. 证明SHMT2过表达可挽救HD模型中的神经退变和运动功能障碍;
5. 发现临床药物氟哌啶醇可通过上调SHMT2恢复组蛋白乳酸化,为HD治疗提供新策略。
亨廷顿病(HD)是一种致命的常染色体显性遗传性神经退行性疾病,由HTT基因CAG重复扩增引起,主要表现为进行性运动功能障碍、认知衰退及纹状体中等多棘神经元(MSNs)的选择性变性。尽管HD的致病基因已明确,但其发病的代谢机制仍不清楚,目前缺乏有效的疾病修饰疗法。
线粒体一碳(mt-1C)代谢是支持核苷酸合成、表观遗传调控和能量产生的核心生化通路,其紊乱与多种神经退行性疾病相关。丝氨酸羟甲基转移酶2(SHMT2)是mt-1C代谢的关键酶,催化丝氨酸转化为甘氨酸,为四氢叶酸循环提供一碳单位。SHMT2功能异常可破坏核苷酸生物合成、氧化还原平衡和表观遗传调控,但其在神经退行性疾病中的作用尚未阐明。
同型半胱氨酸(HCY)是甲硫氨酸循环的关键中间产物,通过叶酸介导的再甲基化与mt-1C代谢偶联。HCY蓄积可导致氧化应激、线粒体功能障碍和DNA甲基化异常,在阿尔茨海默病和帕金森病中已证实与病理特征相关。然而,mt-1C代谢紊乱及HCY在HD中的具体作用机制尚不明确。本研究旨在系统解析SHMT2-HCY轴在HD发病中的作用及其表观遗传调控机制。
代谢组学与转录组学分析:运用靶向代谢组学检测HdhQ7和HdhQ111细胞中256种代谢物,结合RNA测序分析HD患者iPSC来源纹状体类器官(hSOs)的基因表达谱,筛选出一碳代谢紊乱特征。
细胞与类器官模型:建立HD患者iPSC分化的hSOs、HdhQ111/Q7纹状体细胞系及原代MSNs培养体系;采用CRISPRi技术敲低SHMT2,或使用SHIN1药理学抑制SHMT2活性;通过慢病毒和AAV载体实现SHMT2过表达。
动物模型验证:对C57BL/6小鼠纹状体立体定位注射AAV-shSHMT2,或在YAC128 HD小鼠模型中注射AAV-SHMT2,进行转棒、平衡木、旷场等行为学检测。
机制解析:ELISA检测HCY水平;免疫印迹和免疫荧光分析组蛋白乳酸化、神经元标志物及凋亡指标;LiP-MS筛选HCY结合蛋白;分子对接和微量热泳动技术分析HCY与AARS1的相互作用;CUT&Tag联合RNA-seq解析表观遗传调控网络。
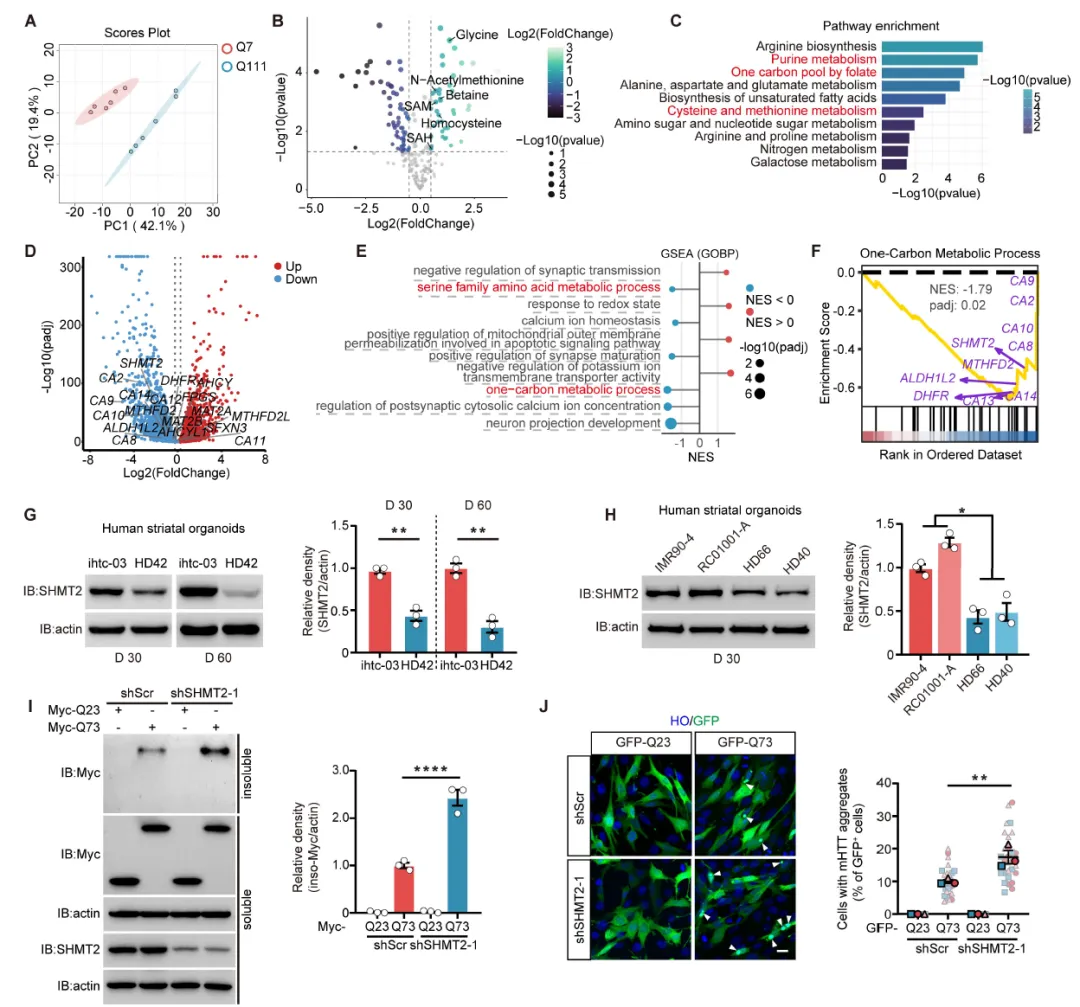
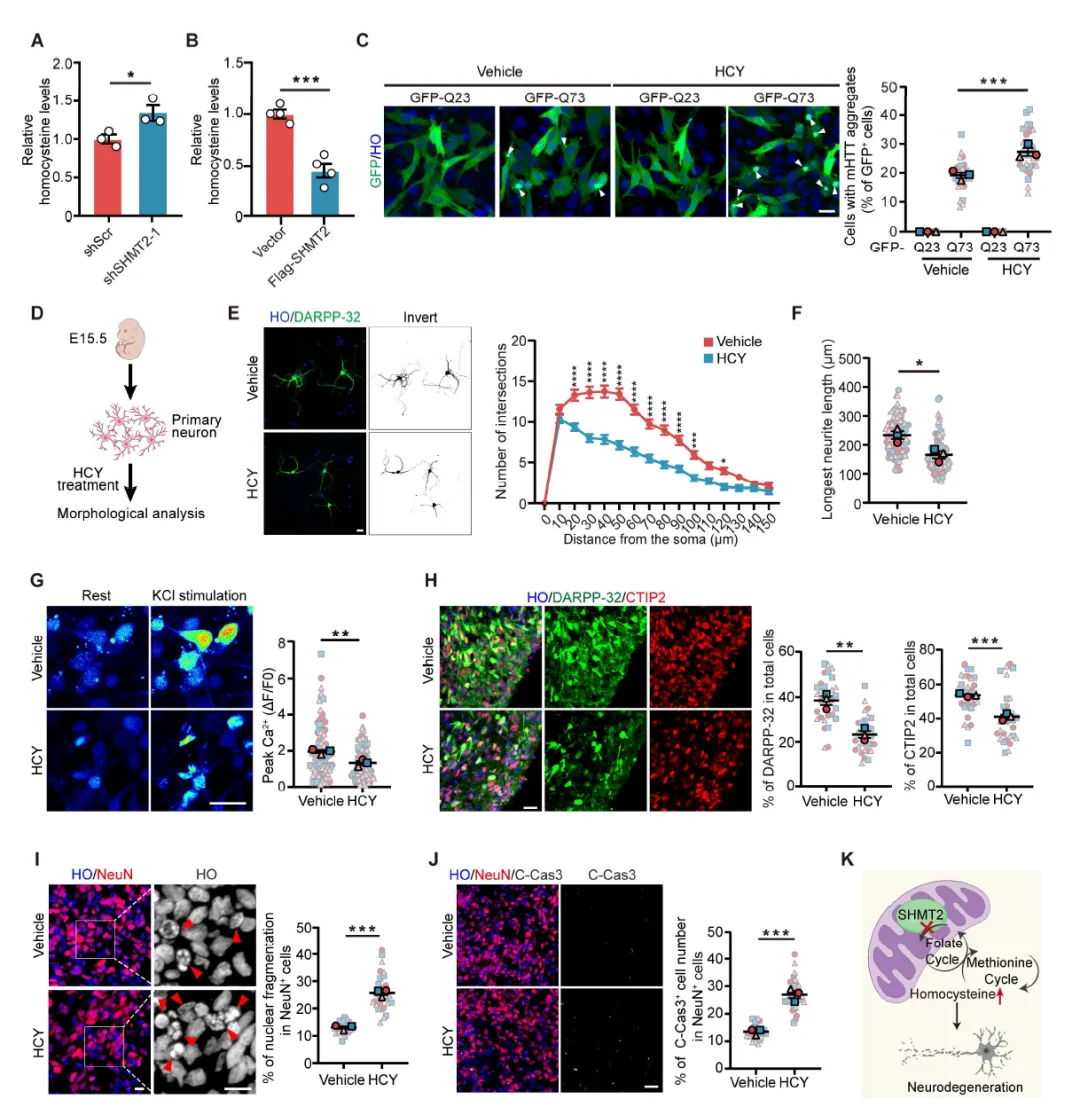
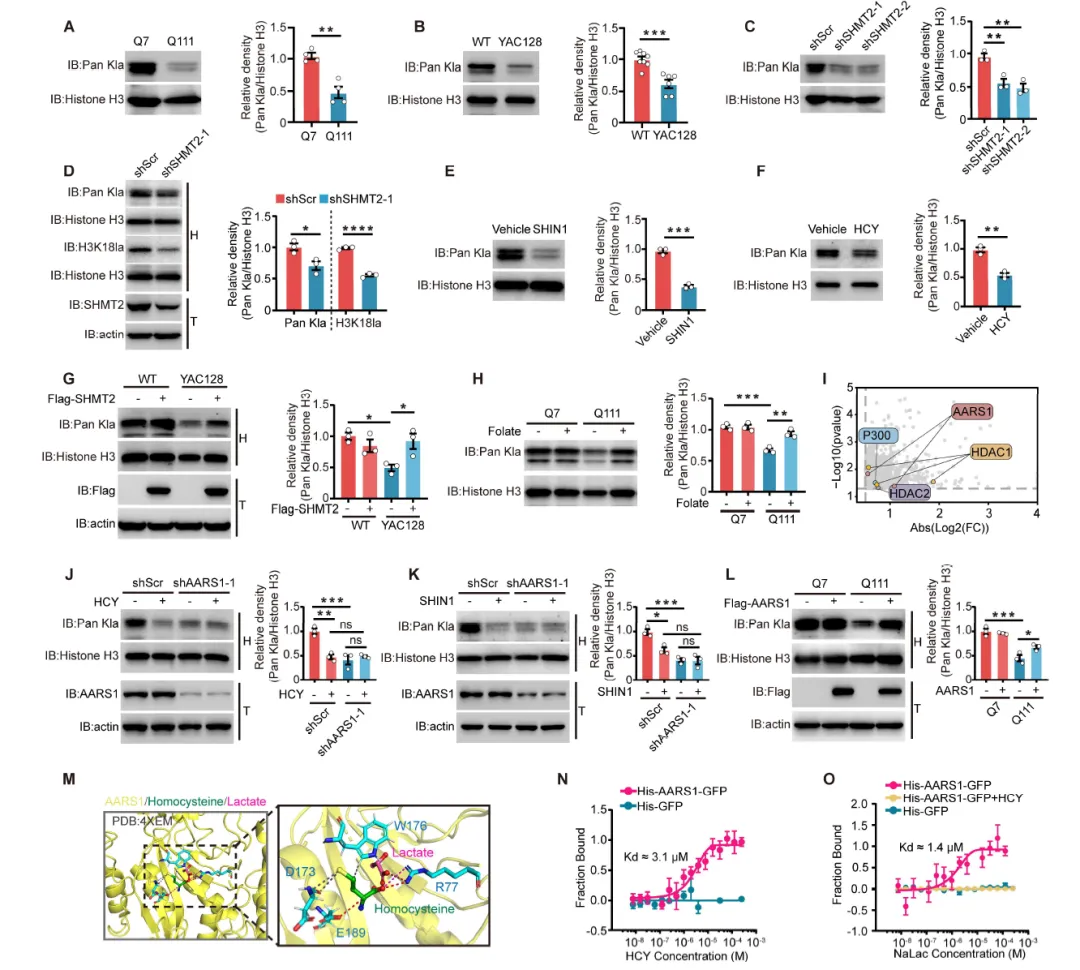
Figure 1:通过代谢组学和转录组学分析,发现HD细胞模型和患者来源纹状体类器官中存在一碳代谢紊乱,表现为同型半胱氨酸、SAM、SAH等代谢物水平升高,SHMT2、ALDH1L2等关键酶基因表达下调;Western blot证实SHMT2蛋白在多种HD模型中显著降低,且SHMT2敲低可增加突变亨廷顿蛋白聚集体的形成。
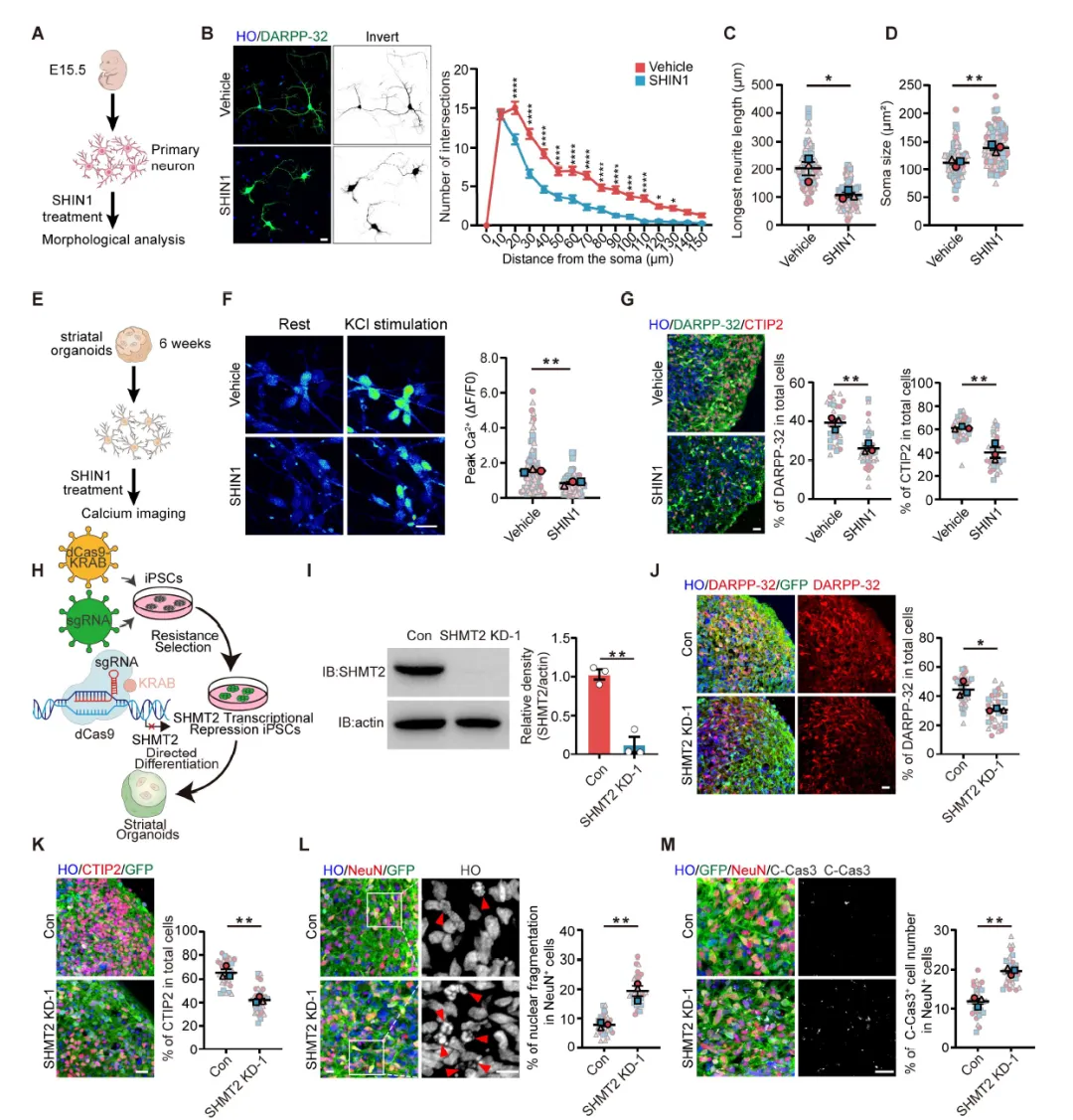
Figure 2:SHMT2抑制或敲低导致原代纹状体神经元和hSOs出现神经退变表型,包括神经突复杂性降低、胞体增大、钙信号响应减弱、DARPP-32+和CTIP2+神经元比例下降,以及cleaved caspase-3阳性凋亡神经元增加,证明SHMT2对维持MSNs结构和功能完整性至关重要。
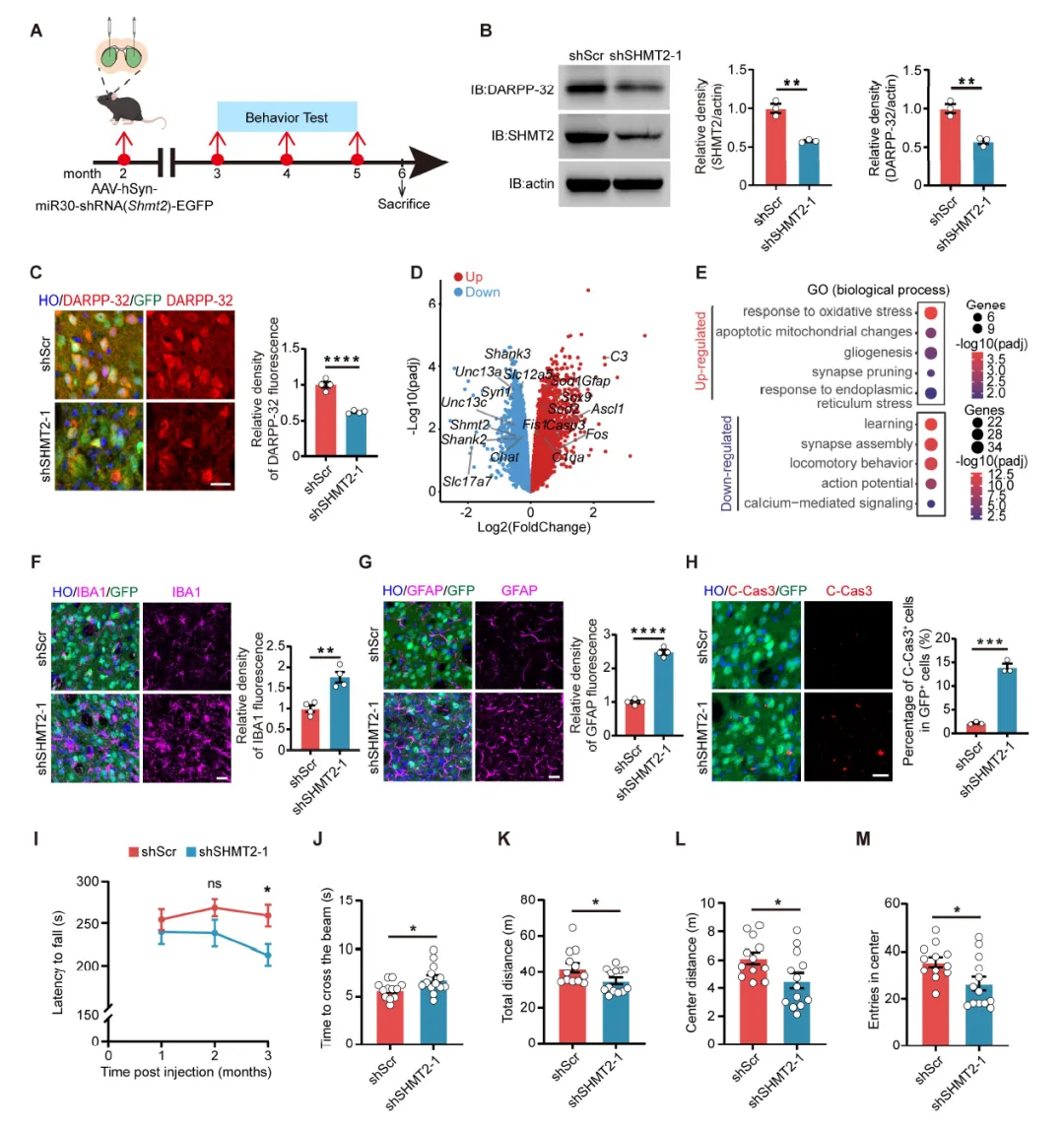
Figure 3:小鼠纹状体注射AAV-shSHMT2后,出现DARPP-32表达降低、氧化应激和线粒体损伤通路激活、突触功能相关基因抑制、星形胶质细胞和小胶质细胞反应性增生、神经元凋亡增加,并在转棒、平衡木和旷场测试中表现出显著的运动协调和探索行为障碍。
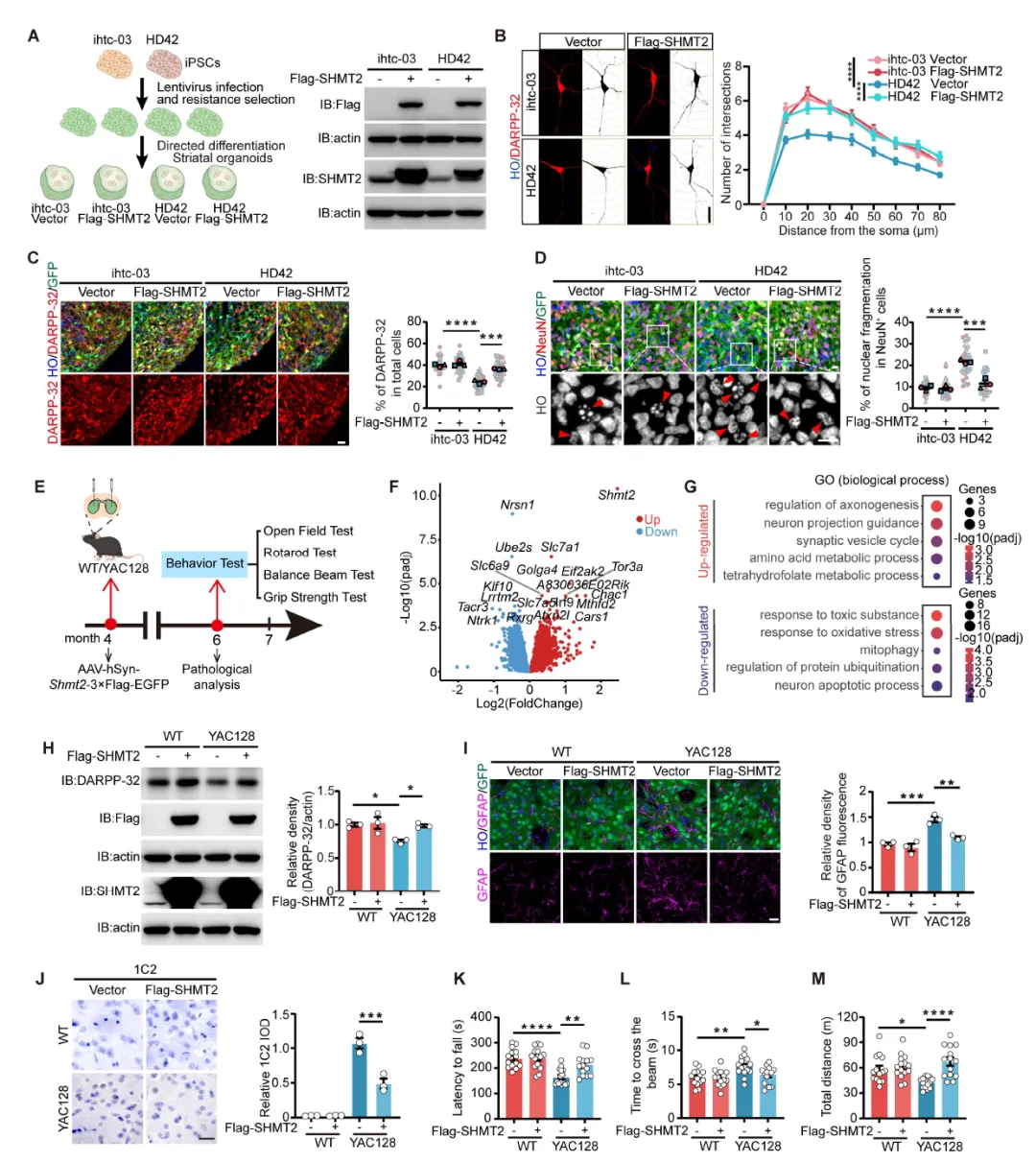
Figure 4:在HD-hSOs中过表达SHMT2可挽救神经突生长缺陷、增加DARPP-32+和CTIP2+神经元比例、减少核碎裂;在YAC128小鼠中,SHMT2过表达可恢复纹状体DARPP-32水平、减轻星形胶质细胞反应性和mHTT聚集、改善运动功能,证明SHMT2具有神经保护作用。
Figure 5:证实SHMT2敲低导致HCY蓄积,而SHMT2过表达可降低HCY水平;外源性HCY处理可增加polyQ聚集体形成、损害神经突生长和钙信号、减少DARPP-32+和CTIP2+神经元、诱导核碎裂和凋亡,表明HCY蓄积是SHMT2缺失介导神经退变的关键机制。
Figure 6:发现HD模型中组蛋白乳酸化水平显著降低,SHMT2敲低、SHIN1处理或HCY处理均可抑制组蛋白乳酸化,而SHMT2过表达或叶酸补充可恢复该修饰;LiP-MS鉴定AARS1为HCY结合蛋白,HCY通过与乳酸竞争性结合AARS1抑制其乳酸转移酶活性,从而调控组蛋白乳酸化。
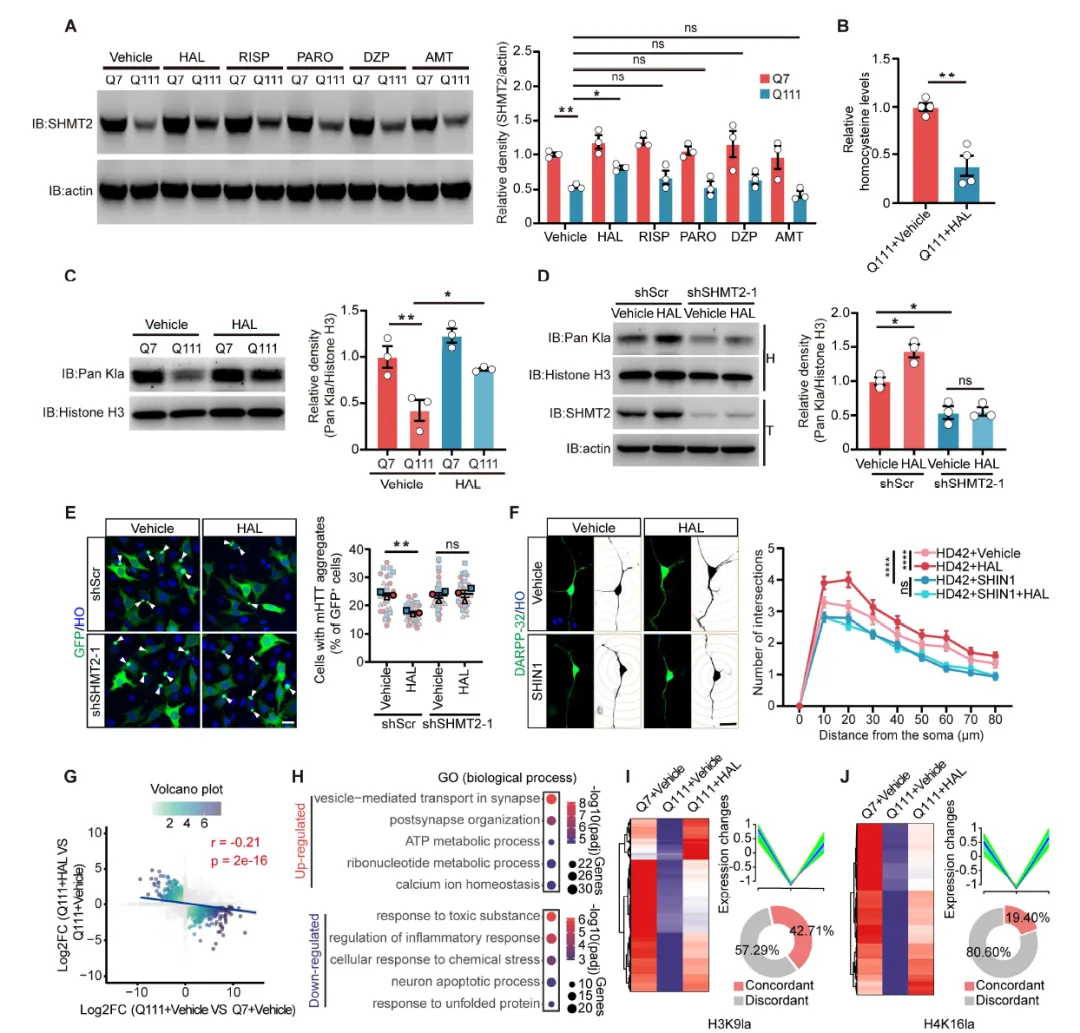
Figure 7:筛选发现氟哌啶醇可上调SHMT2蛋白表达、降低HCY水平、恢复组蛋白乳酸化,该效应依赖于SHMT2;氟哌啶醇可减少polyQ聚集体、改善神经元形态,转录组学和CUT&Tag分析显示其可部分恢复HD相关的基因表达异常和组蛋白乳酸化修饰缺陷。
原文链接
https://pubmed.ncbi.nlm.nih.gov/41805887/
版权声明
标注‘原创’仅代表原创编译,本平台不主张对原文的版权。本平台转载仅出于学术交流和传播信息的需要,不代表本平台观点或证实其内容的真实性。原文版权归原作者所有,作者如不希望被转载或有侵权行为,请联系本平台删除。