南京医科大学,最新Nature Cell Biology!
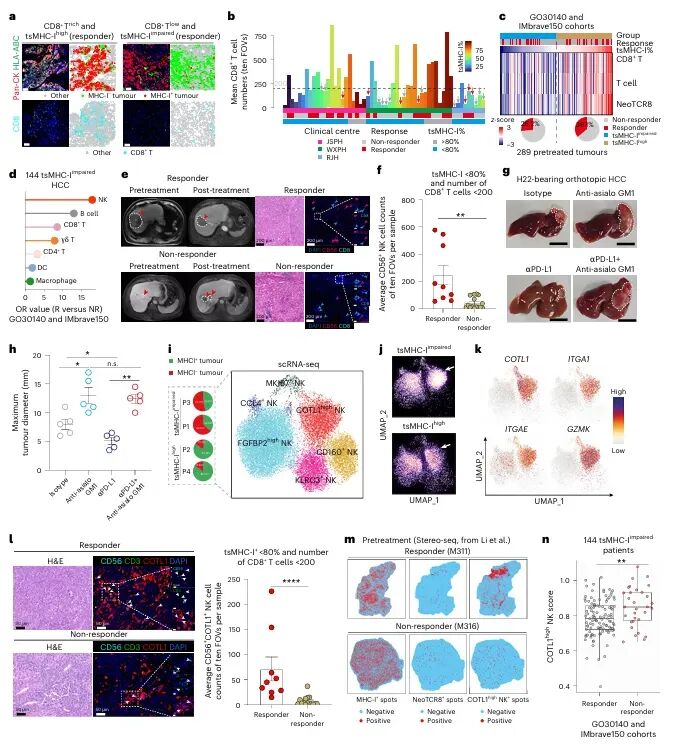
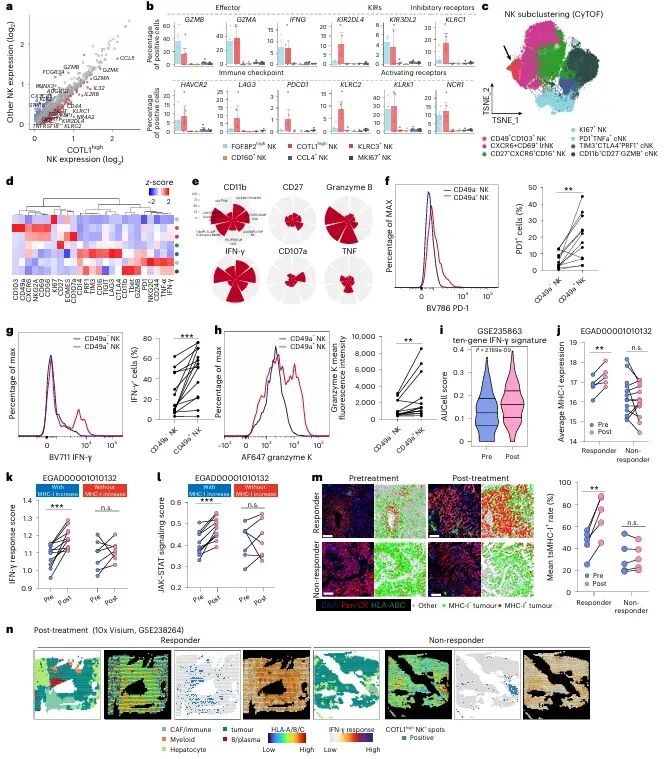
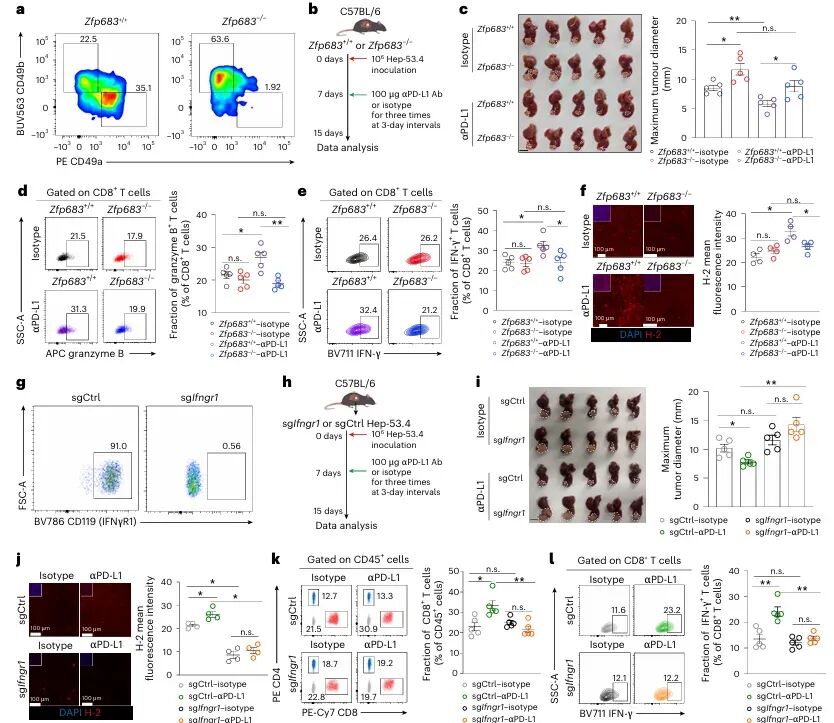
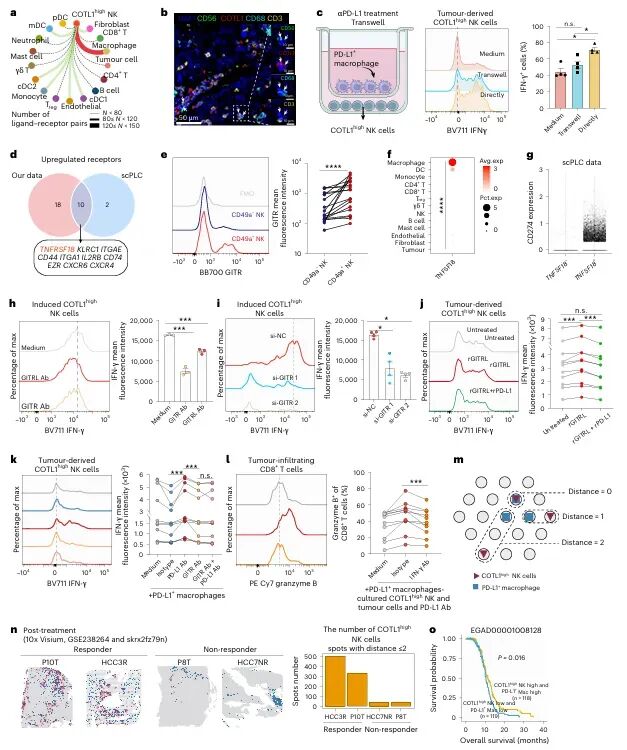
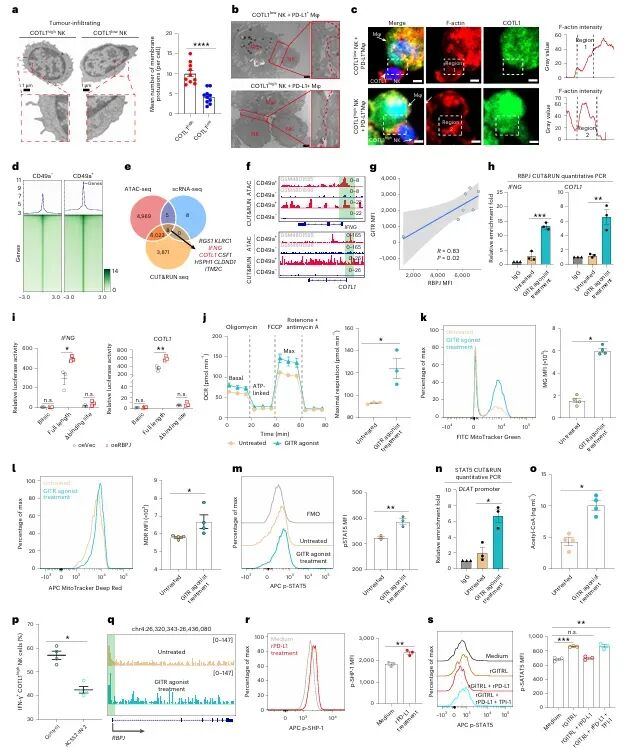
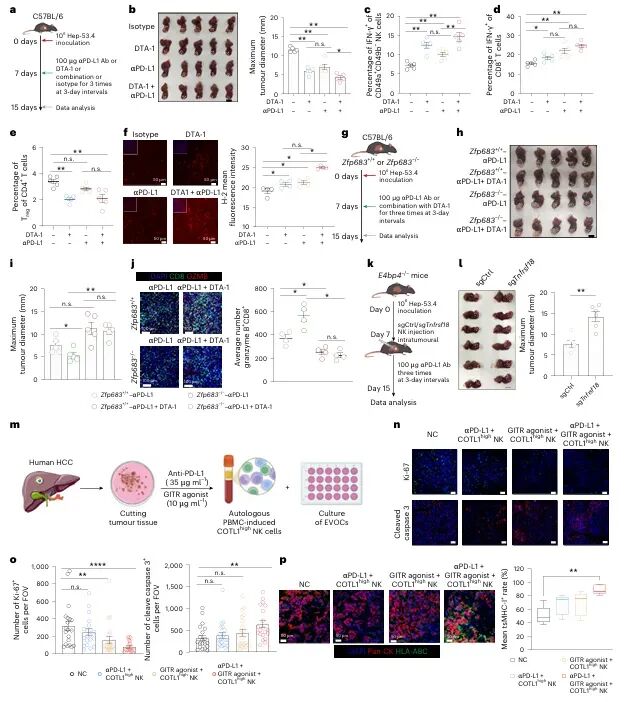
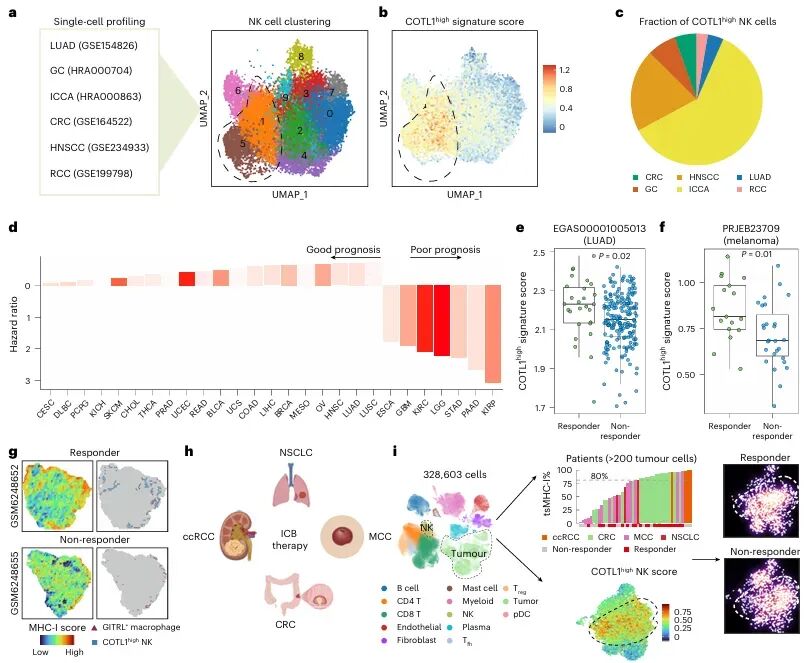
免疫检查点 blockade(ICB)疗法,特别是针对PD-1/PD-L1通路的治疗,已在多种实体瘤中取得了显著成效。然而,临床实践中仍面临巨大挑战:大部分患者无法从该疗法中获益,尤其是在肿瘤特异性主要组织相容性复合体I类分子(tsMHC-I)表达受损的肿瘤中,由于抗原呈递缺陷,CD8⁺ T细胞难以有效识别并杀伤肿瘤细胞,导致免疫治疗响应率极低。尽管如此,临床观察发现,仍有部分tsMHC-I缺陷型肿瘤患者对ICB治疗有响应,其背后的机制尚不明确,成为领域内亟待解决的关键科学问题。近日,南京医科大学陈云团队,中山大学邝栋明,山东第一医科大学种微,以及上海交通大学夏青等人合作,通过系统性研究,发现了一群高表达COTL1的自然杀伤细胞(NK细胞)亚群在介导此类肿瘤的ICB响应中发挥了决定性作用,相关内容以“Reinvigorating COTL1high NK cells via GITR signalling overcomes immune checkpoint blockade resistance in tsMHC-Iimpaired tumours”为题,发表在顶级期刊 Nature Cell Biology 上。图1:预先存在的COTL1high NK细胞与tsMHC-I表达受损及CD8+ T细胞浸润不良肿瘤的免疫治疗疗效相关为探究为何部分tsMHC-I缺陷型肝癌患者仍对ICB治疗有响应,研究者首先对来自多个中心的临床队列进行分析。结果显示,尽管这些患者普遍缺乏CD8+ T细胞浸润,但响应者肿瘤内浸润的NK细胞数量显著高于非响应者(图1e-f)。通过单细胞测序,研究人员在肿瘤浸润NK细胞中鉴定出一个独特的COTL1high亚群(图1i),并且发现该亚群在tsMHC-I缺陷型肿瘤中富集(图1j),尤其在有响应的患者中更为丰富(图1l-n)。这一发现初步确立了COTL1high NK细胞作为预测ICB疗效的关键细胞亚群。图2:肝癌肿瘤中COTL1high NK细胞群的表型特征为了深入了解这群细胞的特征,研究者对其进行了深入的转录组和蛋白水平分析。他们发现,COTL1high NK细胞呈现出一种独特的活化与功能障碍共存的混合状态,既表达GZMK、IFNG等效应分子,也高表达PD-1、TIGIT等耗竭标志物(图2a-b)。蛋白水平验证进一步确认,这群细胞(CD49a+ NK细胞)共表达PD-1、IFN-γ和颗粒酶K(图2f-h)。重要的是,在ICB治疗后,COTL1high NK细胞中的IFN-γ特征评分显著升高(图2i),并伴随着肿瘤细胞MHC-I表达的上调(图2j, m)。这表明,COTL1high NK细胞是ICB治疗后IFN-γ的主要来源,并可能通过IFN-γ促进肿瘤细胞MHC-I的再表达。图3:PD-1/PD-L1阻断增强COTL1high NK细胞产生IFN-γ,从而驱动tsMHC-I表达上调为了证实IFN-γ的关键作用,研究者构建了小鼠原位肝癌模型。他们发现,在Zfp683(控制CD49a+ NK细胞发育的关键转录因子)敲除小鼠中,由于缺乏COTL1high NK细胞,肿瘤生长加速,且抗PD-L1治疗的疗效完全消失(图3c)。同时,在IFN-γ受体敲除的肿瘤细胞中,抗PD-L1治疗也无法诱导MHC-I上调或CD8+ T细胞浸润(图3i-l)。这些结果确凿地证明,COTL1high NK细胞来源的IFN-γ是ICB治疗中促进肿瘤MHC-I表达和CD8+ T细胞活化的关键信号。图4:PD-L1阻断通过GITRL-GITR轴增强COTL1high NK细胞的IFN-γ产生接下来,研究者试图阐明COTL1high NK细胞功能维持的分子机制。细胞互作分析发现,这些NK细胞与PD-L1+巨噬细胞存在密切的相互作用和空间共定位(图4a-b)。在共培养体系中,抗PD-L1抗体能显著增强COTL1high NK细胞的IFN-γ产生,而这种增强作用依赖于细胞间的直接接触(图4c)。进一步分析发现,GITR(由TNFRSF18编码)是COTL1high NK细胞上高表达的一个关键共刺激受体,而其配体GITRL则主要由PD-L1+巨噬细胞表达(图4f)。功能实验证实,阻断GITRL-GITR相互作用会完全消除抗PD-L1抗体诱导的IFN-γ产生(图4h-k)。因此,PD-L1阻断可能通过解除PD-1对GITR信号通路的抑制,从而释放GITRL-GITR介导的NK细胞活化。图5:GITR信号通过代谢-表观遗传调控启动COTL1high NK细胞的IFN-γ产生GITR信号如何增强NK细胞功能?研究者深入探究了其下游机制。他们发现,GITR激活后,COTL1high NK细胞与巨噬细胞形成的免疫突触更为稳定,并且促进了转录因子RBPJ在IFNG和COTL1基因启动子区域的结合(图5h)。同时,GITR激活还显著增强了NK细胞的线粒体氧化磷酸化能力,提高了乙酰辅酶A水平(图5j, o)。进一步研究表明,乙酰辅酶A的增加促进了组蛋白H3K27ac修饰,进而上调了RBPJ的转录(图5q)。这一信号通路形成了一个正反馈循环,即GITR信号通过代谢重编程(增强线粒体功能)和表观遗传修饰(H3K27ac),最终强化了RBPJ介导的IFN-γ和COTL1的转录,从而稳定了免疫突触并维持了NK细胞的效应功能。图6:联合靶向T细胞和COTL1high NK细胞增强tsMHC-I缺陷型肝癌的免疫治疗响应基于上述发现,研究者提出联合GITR激动剂与抗PD-L1可能具有协同抗肿瘤效果。在小鼠模型中,联合治疗相比于单药治疗显著增强了抗肿瘤效果,表现为肿瘤生长抑制、CD49a+ NK细胞和CD8+ T细胞中IFN-γ产生增加,以及肿瘤MHC-I表达上调(图6b-f)。至关重要的是,在Zfp683-/-小鼠或NK细胞特异性敲除GITR的模型中,联合治疗的疗效被完全消除(图6i, l),证明这种协同作用高度依赖于NK细胞自身的GITR信号。此外,在患者来源的肿瘤组织离体培养体系中,联合治疗同样能有效抑制肿瘤增殖并诱导MHC-I表达(图6o-p),为该联合疗法向临床转化提供了强有力的支持。图7:COTL1high NK细胞特征可预测人类MHC-I缺陷型肿瘤的免疫治疗响应最后,研究者将这一发现拓展到更广泛的实体瘤中。他们分析了包括肺癌、黑色素瘤、鼻咽癌等多种肿瘤的公共数据库,发现COTL1high NK细胞样亚群在多种肿瘤中普遍存在,且其丰度与患者生存期正相关(图7a-d)。更重要的是,在tsMHC-I缺陷型肿瘤患者中,对ICB治疗有响应的患者,其肿瘤内COTL1high NK细胞特征评分显著高于无响应者(图7e-f)。这一结果证实了COTL1high NK细胞作为预测ICB疗效的潜在生物标志物,并提示其作用机制可能具有泛癌种意义。综上所述,这项研究首次揭示了在MHC-I缺陷型肿瘤中,一群高表达COTL1的NK细胞亚群是决定ICB治疗成败的关键因素。研究阐明了该细胞亚群如何通过与PD-L1+肿瘤相关巨噬细胞相互作用,利用GITRL-GITR信号轴在PD-L1阻断的背景下被激活,并通过独特的代谢-表观遗传调控回路(线粒体氧化磷酸化-乙酰辅酶A-H3K27ac-RBPJ)维持自身的效应功能,进而产生IFN-γ,重新诱导肿瘤细胞表达MHC-I,并最终“招募”和活化CD8+ T细胞,重塑抗肿瘤免疫。这一机制不仅解释了部分MHC-I缺陷型患者仍能对ICB治疗产生响应的临床现象,更重要的是,它提出了一个极具前景的治疗新策略:联合使用GITR激动剂与PD-1/PD-L1阻断剂,通过“重振”COTL1high NK细胞,有望克服当前免疫治疗在该类“冷肿瘤”中的耐药瓶颈,为广大的tsMHC-I缺陷型肿瘤患者带来新的治疗希望。
「BioMed科技」关注生物医药×化学材料交叉前沿研究进展!交流、合作,请添加杨主编微信!

https://doi.org/10.1038/s41556-026-01925-9来源:BioMed科技声明:仅代表作者个人观点,作者水平有限,如有不科学之处,请在下方留言指正!


 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
