金属活性中心的自旋极化为减轻电催化过程中关键中间体的自旋翻转提供了一种强有力的手段,然而如何有效触发自旋极化并建立其与氧还原反应(ORR)/析氧反应(OER)性能之间的关系仍然具有挑战性。
2026年04月04日,黑龙江大学邹金龙、李丽团队在Advanced Functional Materials期刊发表题为“Selenium Vacancy-Enabled High Spin-Polarized Cobalt Sites to Effectively Mediate Spin Flipping in Oxygen Electrocatalysis”的研究论文,团队成员霍思辰为论文第一作者,邹金龙、南京师范大学付更涛、李丽为论文共同通讯作者。
第一作者:霍思辰
通讯作者:邹金龙、付更涛、李丽
通讯单位:黑龙江大学、南京师范大学
论文DOI:10.1002/adfm.75097
该研究提出了一种缺陷工程方法,在集成到铁单原子平台(FeSA@CoSe₂-Seᵥ)上的二硒化钴(CoSe₂)中引入硒空位(Seᵥ),以增强Co的自旋极化。FeSA@CoSe₂-Seᵥ实现了高ORR半波电位(0.921V)和低OER过电位(370mV@10mA cm⁻²),显著优于FeSA@CoSe₂。当应用于锌-空气电池(ZAB)时,该材料实现了高功率密度(186mW cm⁻²),循环寿命长达582小时。FeSA@CoSe₂-Seᵥ基柔性ZAB即使在0°-180°弯曲条件下也能保持稳定的充放电性能。Seᵥ的引入降低了Co 3d轨道的简并度,有效触发了自旋极化。这种电子结构重构导致Co-*O/*OH的π*轨道失去一个电子,增强了Co 3d与*O/*OH 2p轨道之间的杂化,从而减轻了中间体的自旋翻转。铁磁性FeSA稳定了Seᵥ和Co活性位点,确保了结构/催化稳定性。该研究证实了Seᵥ诱导的自旋极化调控的有效性,为设计双功能电催化剂提供了一种基于自旋电子学的新方法。
随着全球化石能源的枯竭,对清洁、低碳能源的需求日益增长,尤其是在中国,迫切需要新的可再生能源技术。锌-空气电池(ZABs)以其高能量密度和环境友好优势,已成为一种关键的储能技术。氧还原反应(ORR)和析氧反应(OER)在可充电ZABs等能源转换装置中发挥着关键作用。然而,ORR/OER涉及复杂的多电子/质子转移过程,导致难以克服的反应能垒和缓慢的动力学过程。贵金属催化剂(如Pt/C和IrO₂)通常被认为是先进的ORR/OER电催化剂。然而,其有限的可用性和高昂的成本对其大规模应用构成了重大挑战。近年来,过渡金属硫族化合物(TMCs)因其可调的电子结构和在地球上的丰富储量而成为贵金属催化剂的有前景的替代品。然而,其本征半导体特性导致导电性和活性不足,使得ORR/OER活性仍低于贵金属催化剂。与其他硫族元素(氧、硫和碲)相比,硒(Se)具有更高的极化率和金属特性,这赋予过渡金属硒化物独特的电子结构。其中,二硒化钴(CoSe₂)因其本征高导电性和可调活性位点,有利于电子转移并增强催化活性,成为降低OER过电位的理想候选材料。然而,CoSe₂基催化剂在ORR/OER过程中仍面临两大挑战。首先,在碱性环境中结构稳定性不足;其次,ORR和OER的活性不平衡。具体而言,要解决CoSe₂催化剂在ORR/OER中的结构不稳定性和活性不平衡问题,需要对中间体吸附过程的热力学和动力学调控机制进行系统理解。
此前有报道称,金属活性位点与中间体之间的吸附能随d带电子填充状态的变化而变化。这为通过调控自旋和电子结构来优化氧中间体(如*O、*OH、*OOH)的吸附强度提供了理论基础。当金属活性位点发生自旋极化时,其d轨道电子构型发生显著变化,导致自旋向上与自旋向下电子比例的改变。自旋电子占据的差异改变了成键和反键轨道的电子占据数和能级分布,从而调控了金属d轨道与中间体p轨道之间的杂化。这种电子结构的精细调控直接影响中间体的吸附强度,并通过改变反应能垒来控制整个反应动力学。因此,触发自旋极化以优化d-p轨道杂化强度,并确保吸附能处于热力学最优范围内,是提升催化活性的关键机制。此外,在ORR/OER过程中,OH⁻/H₂O与O₂之间的自旋转变也对反应动力学有关键影响。具体而言,OH⁻/H₂O是自旋单重态,所有电子均配对,没有未配对电子;而基态O₂分子是自旋三重态(³O₂),在两个简并π*轨道(πₓ*和πᵧ*轨道)中各有一个未配对电子。根据自旋守恒原理,反应物和产物的总自旋态必须保持一致。因此,OH⁻/H₂O的自旋单重态与非磁性表面上的自旋三重态O₂直接相互作用会经历自旋阻塞效应,需要更高的自旋翻转能垒才能进行吸附和活化。这要求催化剂产生自旋极化电子并创建有效的自旋电子转移通道,促进电子转移过程中的自旋翻转,以克服自旋阻塞效应,实现OH⁻/H₂O与O₂之间的自发转换。
空位工程作为一种自旋工程策略,可以调控金属活性位点的电子自旋态,触发自旋极化,从而影响催化剂与反应物分子之间的轨道重叠和电荷转移路径。当前研究中研究最广泛的空位类型是阴离子空位和阳离子空位,其中阴离子空位由于其较低的形成能而更容易形成。阴离子空位可以增强自旋极化,增加费米能级附近的电子态密度,降低电荷转移电阻,并优化关键中间体的吸附/脱附,所有这些都显著提高了催化性能。例如,Wang等人将非晶态RuO₂引入NiO中以降低阴离子空位的形成能,促进了氧空位(Oᵥ)的产生。因此,尿素氧化反应的催化反应能垒显著降低。Si等人发现表面活性剂甲基三苯基溴化膦的腐蚀作用导致介孔MnO₂纳米片中部分Mn-O键断裂,导致Oᵥ的形成,进而促进了高自旋极化Mn³⁺-Mn³⁺对的形成。Mn的d轨道与N₂轨道之间的双向电子转移增强了Mn³⁺-Mn³⁺对的自旋极化程度,为降低氮还原反应的能垒提供了关键条件。这些观察结果表明,空位工程可以破坏金属活性位点的配位环境和电子结构对称性,诱导电子自旋极化。尽管关于Oᵥ的研究已取得一些进展,但关于阴离子空位,特别是Seᵥ,如何通过各种电催化过程中的自旋极化来优化原始的原子间相互作用平衡和本征催化活性的系统性研究尚未见报道。因此,对于上述CoSe₂,深入阐明Seᵥ与CoSe₂中自旋极化之间的内在相互作用机制,并明确ORR/OER中间体的吸附机制具有很高的价值。
在此,该研究首次采用缺陷工程策略,将Seᵥ引入集成在铁磁性Fe单原子(FeSA)基底上的CoSe₂中,成功构建了FeSA@CoSe₂-Seᵥ复合电催化剂。FeSA具有多自旋态特性和高导电性,可以作为稳定的基底材料,为赋予宏观铁磁性提供基础铁磁平台,并为自旋极化创造自旋有序的微环境。Seᵥ的引入破坏了周围Co原子的电荷平衡,导致邻近Co位点发生电子重新分布,并增强了自旋极化。磁性实验和理论计算表明,Seᵥ可以触发CoSe₂中Co位点的电子离域。具体而言,低能量的Co 3d轨道向高能量轨道注入一个电子,导致费米能级附近自旋向上和自旋向下电子态分布的不平衡。这诱导了高自旋极化和增强的磁性,最终驱动Co物种的自旋构型从中自旋(IS)态转变为高自旋(HS)态。基于分子轨道理论(MOT),半填充的dz²轨道与氧中间体(*OH和*O)的2pz轨道杂化形成σ键,有效优化了中间体的吸附能。因此,自旋极化的FeSA@CoSe₂-Seᵥ催化剂表现出优异的ORR/OER活性,使其在水系和柔性ZABs中能够实现卓越的性能。该研究为自旋极化与催化性能之间的构效关系提供了新见解,并为设计高效的双功能催化剂提供了理论指导。
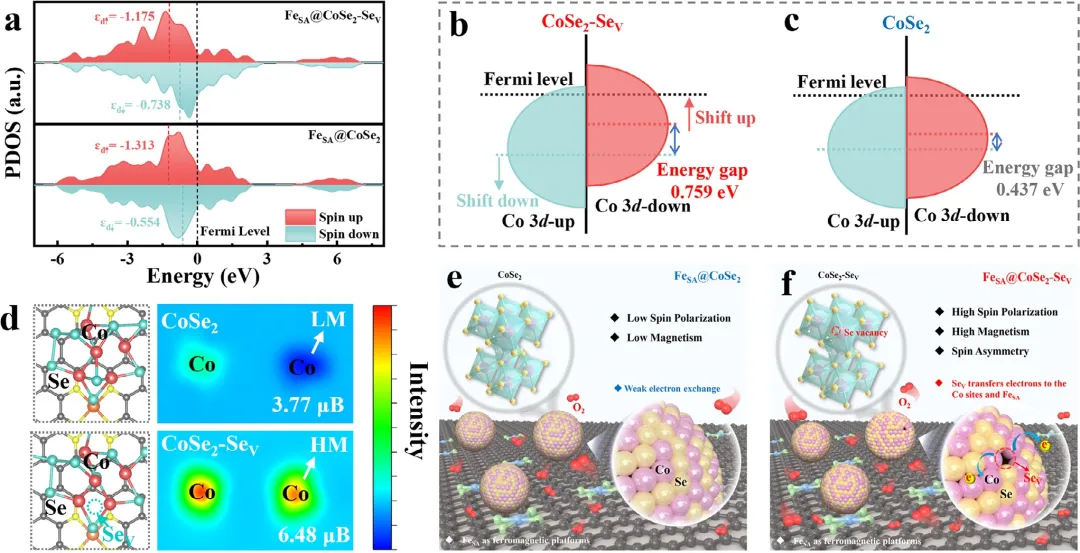
图1 (a) FeSA@CoSe₂和FeSA@CoSe₂-Seᵥ的PDOS(费米能级和d中心(εd)用虚线表示)。(b) FeSA@CoSe₂-Seᵥ中CoSe₂-Seᵥ和(c) FeSA@CoSe₂中CoSe₂的Co 3d自旋极化示意图。(d) FeSA@CoSe₂(上)和FeSA@CoSe₂-Seᵥ(下)的优化结构构型和计算出的二维自旋密度图。(e) 低自旋极化的FeSA@CoSe₂和(f) 高自旋极化的FeSA@CoSe₂-Seᵥ的自旋极化差异示意图。
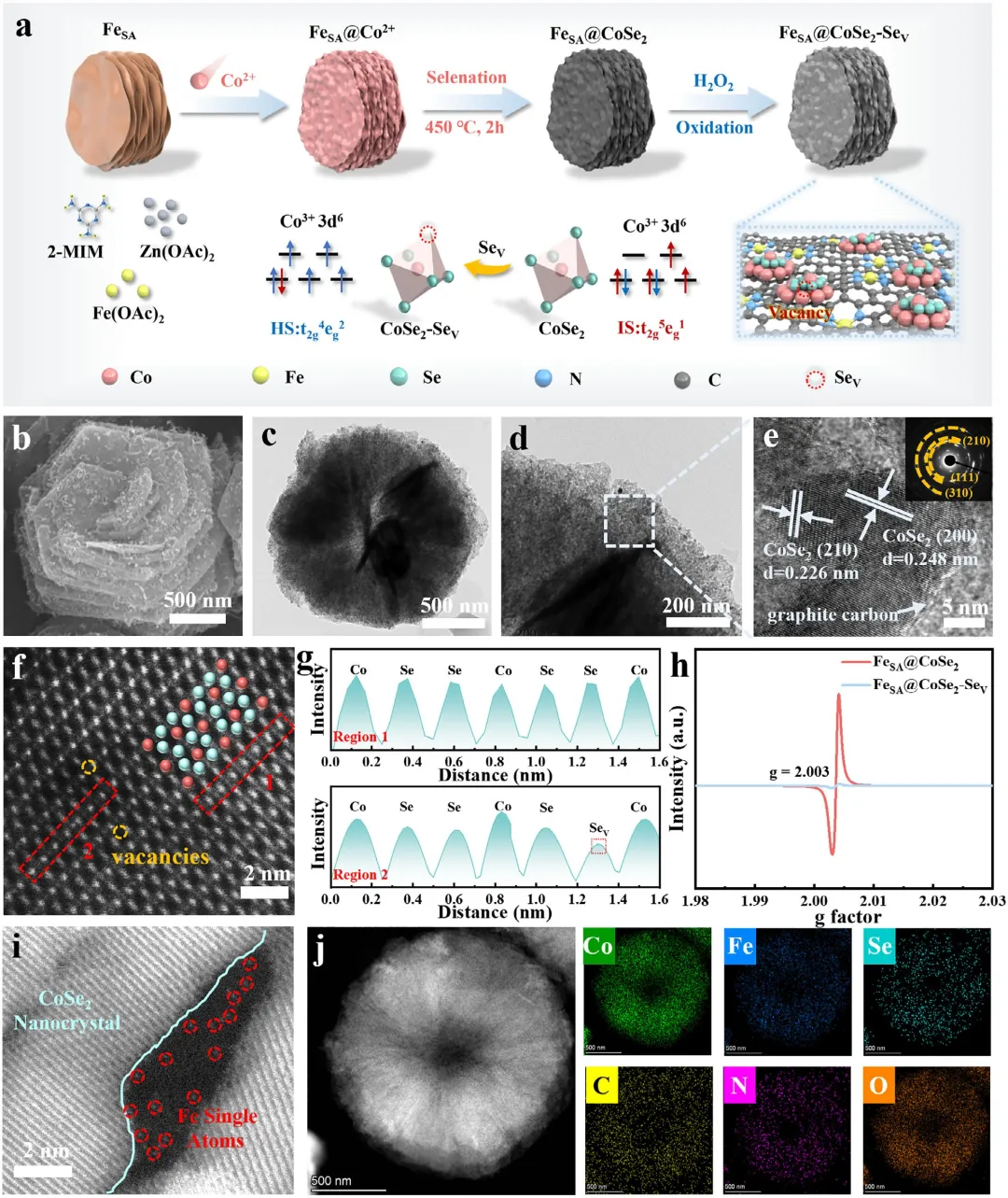
图2 (a) FeSA@CoSe₂-Seᵥ的合成示意图。(b) FeSA@CoSe₂-Seᵥ的SEM图像。(c,d) TEM图像和(e) HRTEM图像及SAED图(插图)。(f,g) (210)晶面的HAADF-STEM图像及选定区域1和2的强度图;在原子排列中,红色原子代表Co原子,蓝色球体代表Se原子。(h) FeSA@CoSe₂-Seᵥ和FeSA@CoSe₂的EPR图谱。(i) FeSA@CoSe₂-Seᵥ的像差校正HAADF-STEM图像。(j) FeSA@CoSe₂-Seᵥ中Co、Fe、Se、C、N和O的HAADF-STEM图像及元素面分布图。
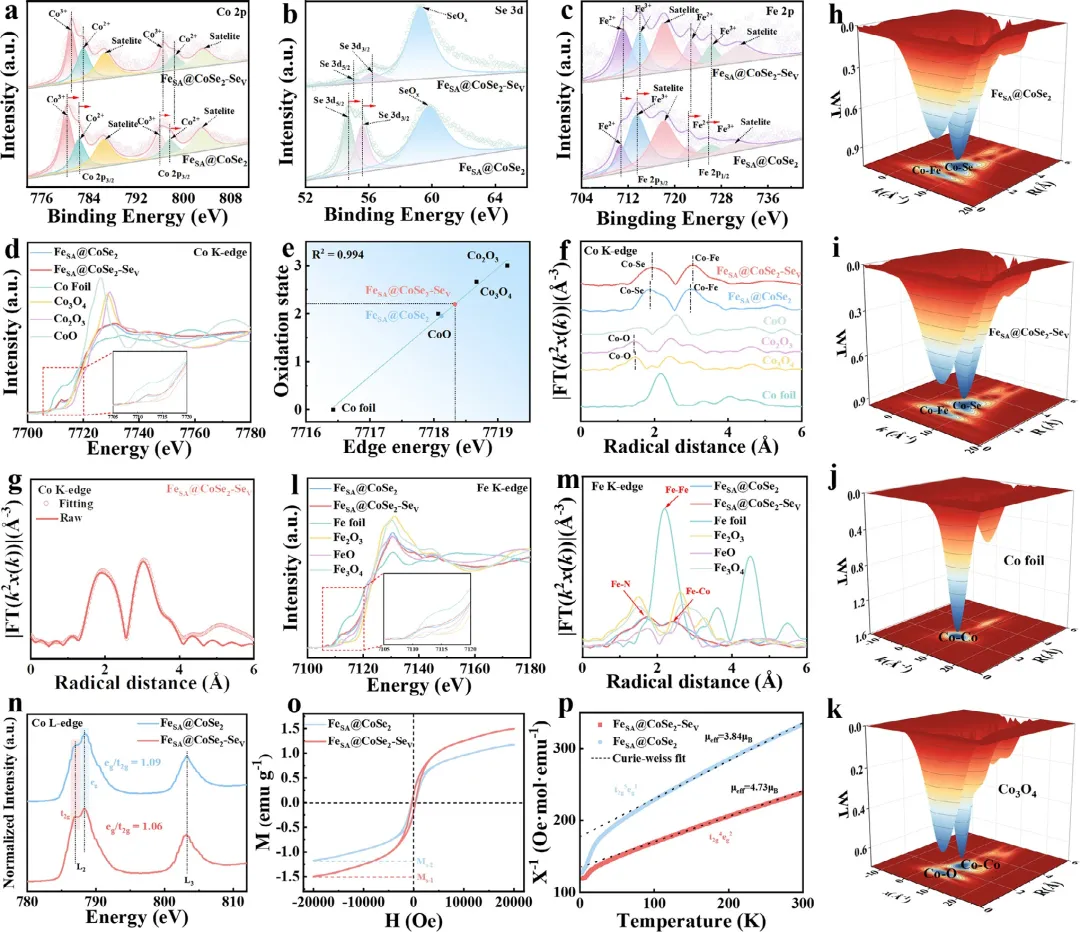
图3 (a) Co 2p、(b) Se 3d和(c) Fe 2p的高分辨XPS谱图。(d) FeSA@CoSe₂-Seᵥ、FeSA@CoSe₂、Co箔、CoO、Co₂O₃和Co₃O₄的Co K-edge XANES谱图。(e) 根据相应的Co K-edge XANES谱图线性拟合得到的Co价态。(f) Co K-edge的FT-EXAFS拟合曲线。(g) FeSA@CoSe₂-Seᵥ在R空间的Co K-edge EXAFS拟合结果。(h) FeSA@CoSe₂、(i) FeSA@CoSe₂-Seᵥ、(j) Co箔和(k) Co₃O₄的k²加权Co K-edge EXAFS信号的小波变换。(l) Fe K-edge XANES谱图。(m) Fe K-edge的FT-EXAFS拟合曲线。(n) FeSA@CoSe₂-Seᵥ和FeSA@CoSe₂的Co L-edge谱图。(o) 10 K时磁化强度与磁场关系(M-H)曲线。(p) χ⁻¹-T曲线及有效磁矩(μeff)。
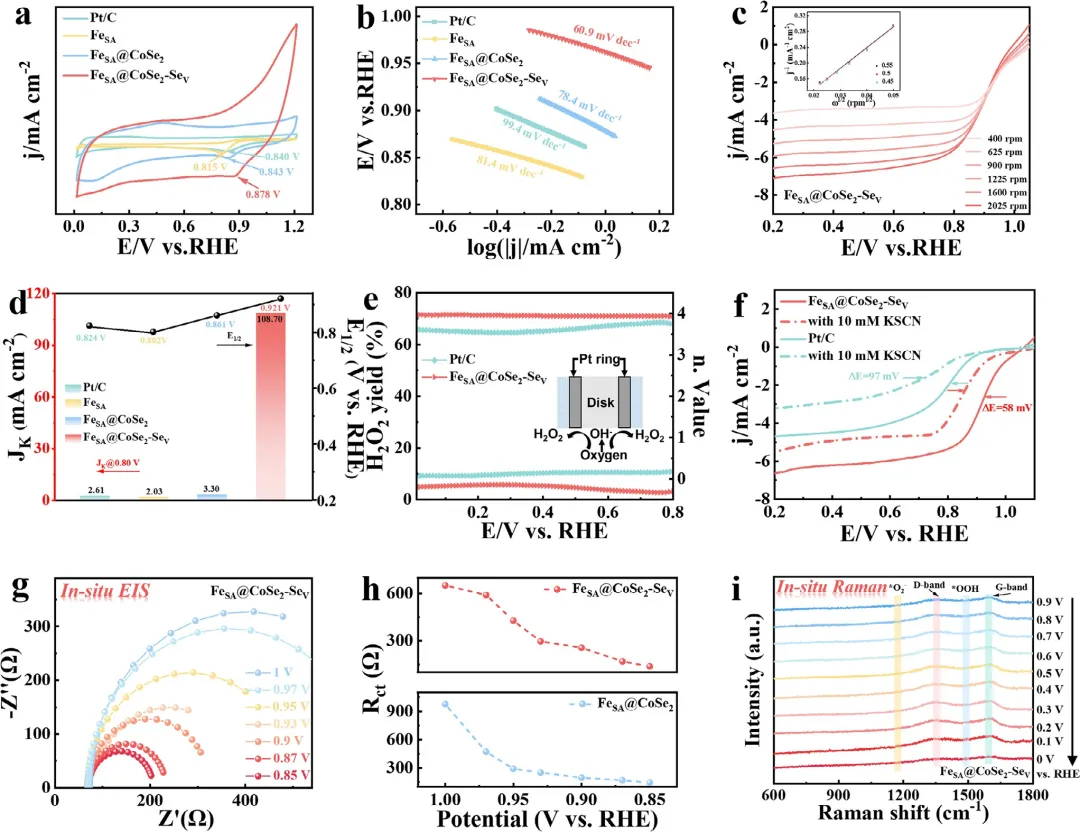
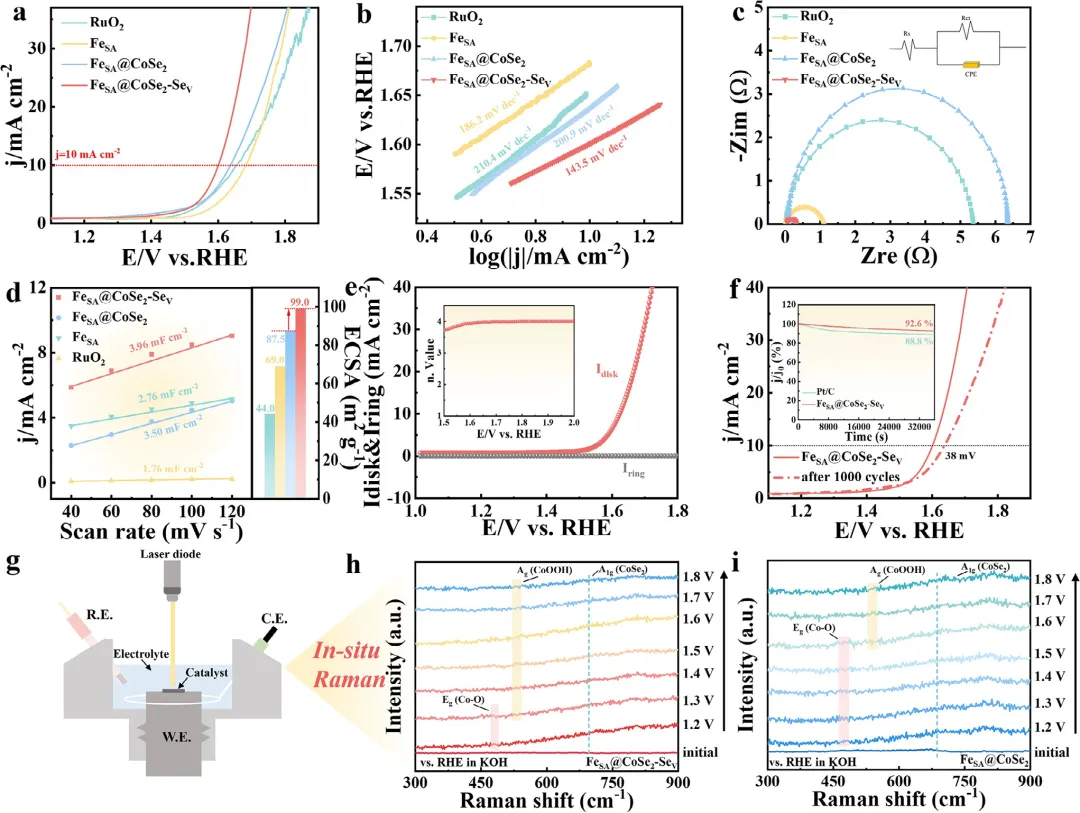
图4 (a)FeSA、FeSA@CoSe₂、FeSA@CoSe₂-Seᵥ和Pt/C的ORR极化曲线。(b) Tafel图。(c) FeSA@CoSe₂-Seᵥ在不同电位下的LSV曲线及计算得到的K-L图(插图)(5 mV s⁻¹)。(d) 各种催化剂的E₁/₂和Jₖ。(e) H₂O₂产率和n值。(f) FeSA@CoSe₂-Seᵥ在加入10 mM KSCN前后于0.1 M KOH中的LSV曲线。(g,h) FeSA@CoSe₂-Seᵥ的原位奈奎斯特图及相应的氧物种吸附电阻。(i) FeSA@CoSe₂-Seᵥ在ORR过程中收集的原位拉曼光谱。
图5 (a) LSV曲线(2 mV s⁻¹和1600 rpm),(b) Tafel图,以及(c) 在5 mV振幅和1600 rpm条件下的奈奎斯特图。(d) 催化剂的Cdl(左)和ECSA值(右)。(e) RRDE伏安图(插图为相应的n值)。(f) FeSA@CoSe₂-Seᵥ在1600 rpm连续运行前后的OER极化曲线(插图为计时电流(i-t)响应曲线)。(g) 用于拉曼表征的原位池示意图。(h,i) 在OER过程中收集的FeSA@CoSe₂-Seᵥ和FeSA@CoSe₂的原位拉曼光谱。
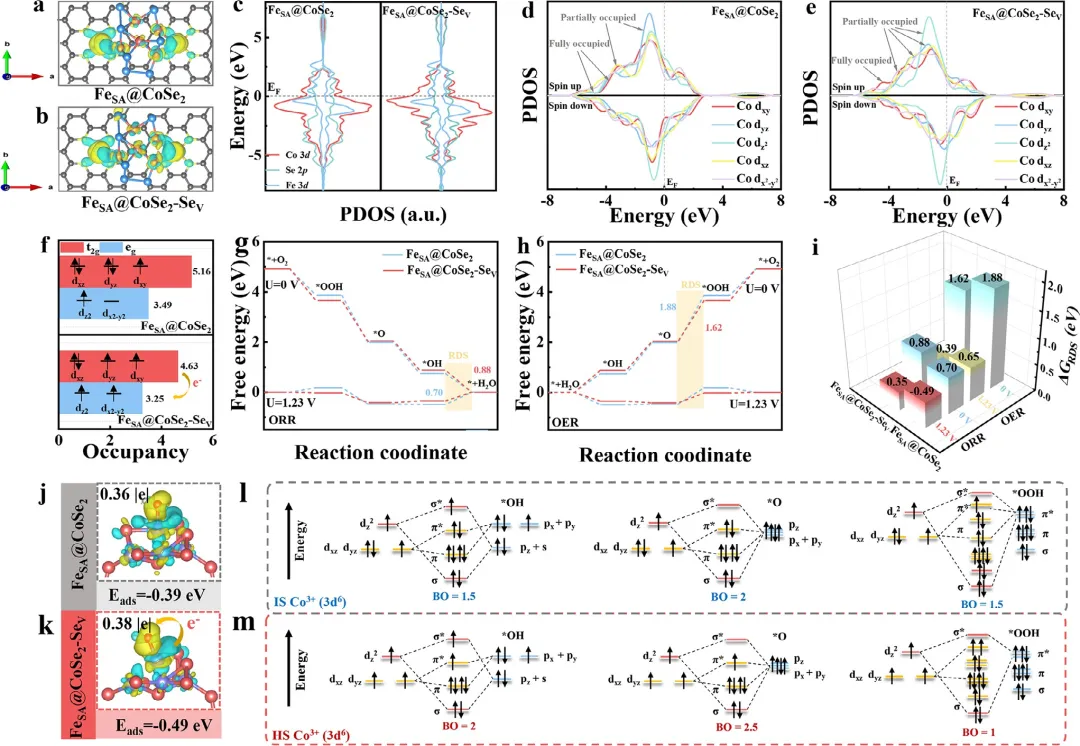
图6 (a,b) FeSA@CoSe₂和FeSA@CoSe₂-Seᵥ复合结构的差分电荷图(黄色和蓝色区域分别代表电子积累和耗尽)。(c) FeSA@CoSe₂和FeSA@CoSe₂-Seᵥ的PDOS。(d,e) FeSA@CoSe₂和FeSA@CoSe₂-Seᵥ的Co 3d轨道PDOS。(f) 计算得到的Co 3d轨道占据数。(g) ORR和(h) OER在零电位(U=0)和平衡电位(U=1.23V)下的自由能图。(i) 计算得到的*OH吸附能和*O脱附能。(j) FeSA@CoSe₂和(k) FeSA@CoSe₂-Seᵥ上吸附*OH的电荷密度差。(l) FeSA@CoSe₂和(m) FeSA@CoSe₂-Seᵥ上*OH、*O和*OH的轨道相互作用。
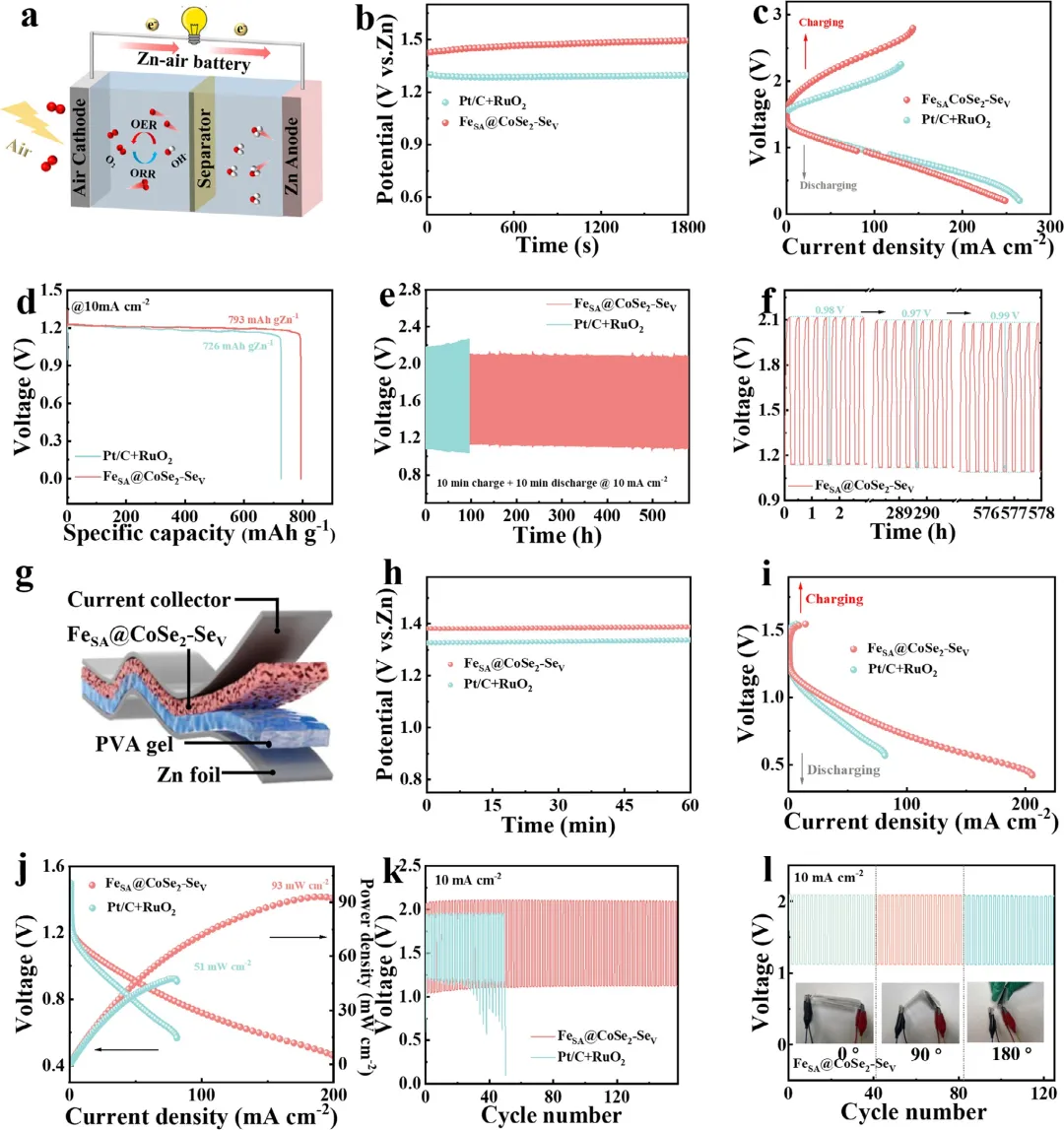
图7 (a) 自制ZAB的示意图。(b) 使用不同催化剂的ZAB的开路电压(OCV)图。(c) 充放电极化曲线。(d) ZAB在10 mA cm⁻²下的放电曲线。(e) 长期恒流充放电循环图。(f) ZAB的往返效率。(g) FZAB示意图。(h) OCV图。(i) 充放电极化曲线。(j) 放电极化曲线及相应的功率密度图。(k) FZAB的循环性能。(l) 采用FeSA@CoSe₂-Seᵥ阴极的FZAB在不同弯曲角度下的循环测试。
总之,该研究通过刻蚀处理成功将Seᵥ引入CoSe₂中,有效调控了CoSe₂的自旋极化程度并提高了其ORR/OER动力学。所制备的FeSA@CoSe₂-Seᵥ催化剂表现出优异的双功能性能,ORR的起始电位为0.986V,OER的电荷转移电阻(Rct)低至0.3Ω。当应用于水系和柔性电池时,FeSA@CoSe₂-Seᵥ表现出高开路电压和优异循环耐久性,优于商业催化剂。理论计算和机理分析表明,Seᵥ通过诱导Co活性位点周围的电荷积累和Co 3d轨道的电子重排,显著促进了高自旋极化的形成。因此,由此产生的高自旋极化Co³⁺优化了催化剂与中间体之间的d-p轨道杂化强度,从而获得了优异的双功能性能。此外,铁磁性FeSA载体可以有效稳定Seᵥ和活性位点,为催化反应的持续稳定进行提供了保障。这不仅增强了与*OH和*O的结合以加速中间体的转化,还减弱了与*OH的相互作用以促进氧气脱附,协同改善了ORR/OER动力学。该研究揭示了缺陷诱导的自旋极化与催化活性之间的内在关联,为设计高效电催化剂提供了重要的理论和实验参考。