单原子催化剂(SACs)已成为在各种催化反应中实现优异活性和选择性的有力候选材料,然而其合成通常需要复杂的技术或还原剂来抑制金属原子聚集。
2026年04月11日,南京工业大学邢卫红、仲兆祥、郑建忠团队在Advanced Functional Materials期刊发表题为“Aprotic Solvent-Mediated Microenvironment Engineering for Single-Atom Catalysts Enables Selective Nitroarene Hydrogenation”的研究论文,南京工业大学Han Xiaozheng、Duan Pengjun为论文共同第一作者,邢卫红、仲兆祥、郑建忠为论文共同通讯作者。
第一作者:Han Xiaozheng、Duan Pengjun
通讯作者:邢卫红、仲兆祥、郑建忠
通讯单位:南京工业大学
论文DOI:10.1002/adfm.75375
该研究报道了一种使用丙酮作为非质子溶剂直接合成原子分散Au₁/COF SACs的简便方法,该溶剂能够将Au前驱体直接锚定到亚胺键合共价有机框架(COFs)上。随后系统调控溶剂组成和前驱体浓度,以实现从单原子到纳米颗粒的Au物种的可控合成。Au₁/COF SAC实现了2.53 wt.%的高Au载量,并在对硝基苯酚加氢反应中表现出优异的周转频率(5000 h⁻¹),以及在多种硝基芳烃加氢反应中的高选择性。对比实验和密度泛函理论DFT计算表明,丙酮的弱给质子能力促进了Au前驱体的逐步脱氯,通过COFs中亚胺位点的质子化形成稳定的Au—N键。此外,Au单原子位点有利于优先结合—NO₂基团,显著提高了硝基芳烃加氢反应选择性。该研究为SACs提供了一种简单的合成策略,并为先进的COF基催化系统提供了一种绿色且节能的方法,具有工业应用潜力。
随着全球对高附加值产品需求的持续增长,苯胺及其衍生物作为重要的有机中间体,在制药、农药和石油化工中扮演着关键角色。据统计,2023年全球苯胺产量达到约990万吨,预计到2030年将增至约1400万吨。目前,超过80%的工业苯胺生产依赖于催化加氢过程,该过程涉及硝基苯与氢气在极端条件下(温度150–200°C,压力2–5 MPa)反应。然而,该过程能耗高、存在一定风险,并产生大量含有硝基苯和苯胺的废水。因此,在更温和、环境友好的条件下寻找高效、高选择性的硝基芳烃加氢催化剂,仍然是实现环境保护和资源再利用双重目标的一项重大挑战。
单原子催化剂(SACs)作为一类有前景的催化材料正在兴起,其特点是金属原子以原子级分散形式负载在各种基底上。这种独特的构型可实现近100%的金属原子利用率、明确的配位环境和独特的电子特性,从而在加氢、氮还原、一氧化碳氧化和二氧化碳还原等关键转化反应中展现出卓越性能。然而,SACs的一个关键局限性在于,由于孤立金属原子的尺寸小和表面能高,在制备和反应过程中容易聚集并失活。这种聚集是由最小化表面积的热力学趋势驱动的;具有较高表面能的较小颗粒本质上不稳定,并且容易聚结成较大的、能量较低的结构。尽管已经研究了包括碳材料、金属氧化物、分子筛和金属有机框架(MOFs)在内的众多载体材料来解决该问题,但精确控制金属原子的配位环境和电子结构仍然是一项艰巨的挑战。
共价有机框架(COFs)作为通过有机单体可逆共价键合组装而成的结晶多孔材料,在气体存储、催化、传感和分离领域引起了广泛兴趣。作为极具前景的单原子金属载体,COFs提供了可预先设计的孔径和通道几何形状,能够有效限域单原子金属,从而抑制金属聚集并精确调控其配位环境。其结构确保了框架中的结合位点(如亚胺氮原子)在整个材料中均匀分布,避免了非晶态载体中常见的不均匀位点分布,这对于实现一致的催化性能至关重要。此外,许多COFs表现出优异的化学稳定性,在苛刻条件(酸性、碱性、高温)下保持稳固的结构完整性,并允许通过化学修饰来增强金属-载体相互作用。尽管已经开发了多种将金属单原子负载到COFs上的策略,主要包括:(1) 在COFs缩合反应过程中,使用强配位配体(如卟啉和酞菁)进行同步锚定;(2) 将金属前驱体与预先合成的COFs孔道内的结合位点配位,随后还原;(3) 用杂原子(如N、S、P)对COFs进行功能化修饰以提高锚定效率。然而,这些方法常常受到反应条件苛刻、功能化后可能改变孔道结构以及负载效率低(尤其是对于贵金属单原子)的困扰。因此,开发一种简单、高效且广泛适用的策略,用于在COFs上有效限域并精确控制贵金属单原子的粒径,仍然是一项重大挑战。
基于此,该研究开发了一种通过丙酮浸渍合成原子分散的Au₁/COF SACs的简便策略,这是首次尝试通过调控溶剂在COFs上可控制备单原子催化剂。丙酮作为一种非质子溶剂,具有低给质子和低接受质子的特性,能够实现对亚胺键合COFs的可控质子化,从而使Au单原子高度分散。密度泛函理论DFT计算表明,丙酮创造的微环境促进了Au前驱体的逐步脱氯,通过与质子化的亚胺位点配位形成稳定的Au—N键。此外,孤立的Au单原子优化了局域电子环境,降低了硝基芳烃加氢的活化能垒,从而增强了反应动力学和底物选择性。因此,所得催化剂在对硝基苯酚加氢中表现出卓越的性能,并在多种硝基芳烃中保持高选择性(>99.9%)。该研究展示了一种通过溶剂控制来调控COF负载SACs中金属颗粒尺寸和电子结构的绿色策略,最终提升了催化性能。
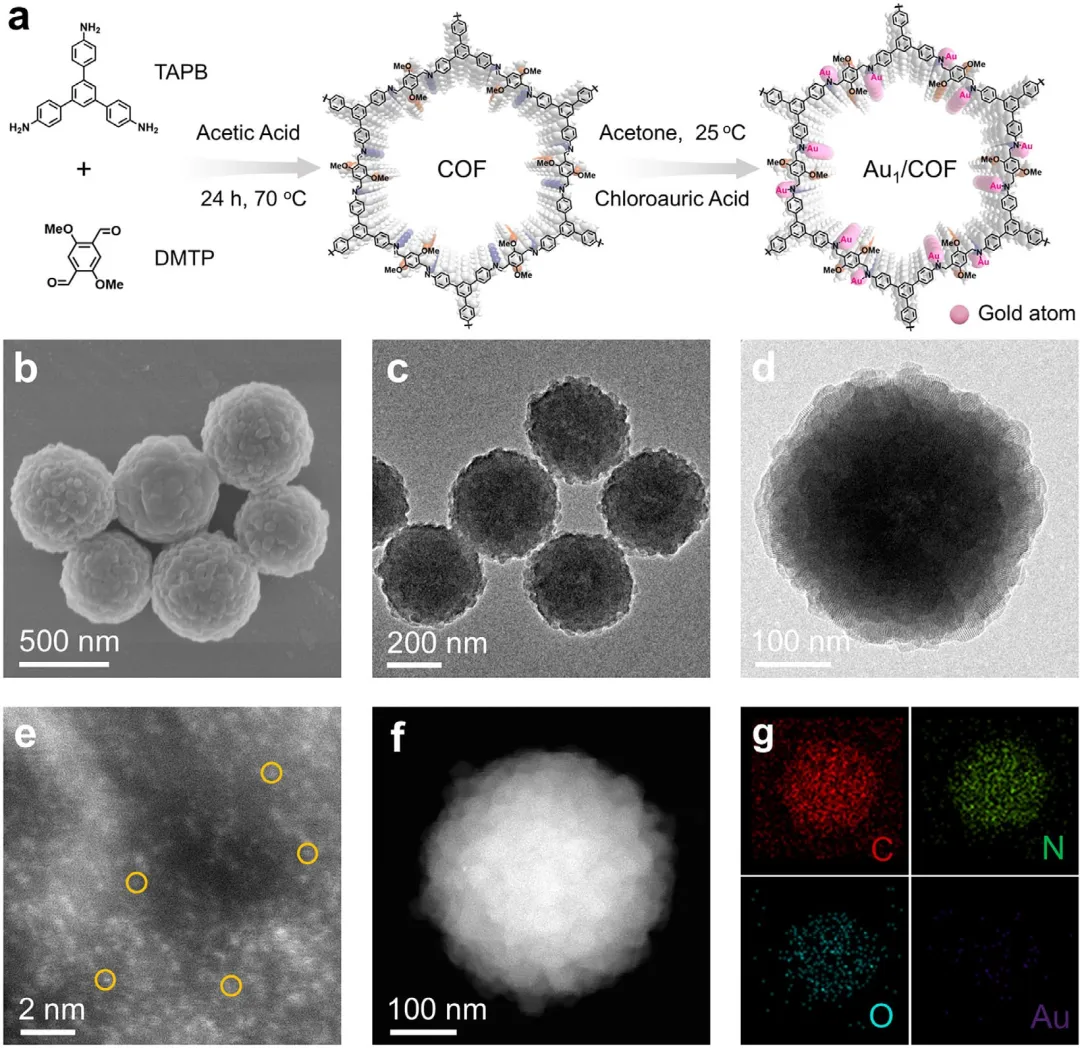
图1 | 催化剂的合成路线和形貌结构。(a) Au₁/COF复合材料的合成路线示意图。(b) 所制备Au₁/COF的SEM图像。(c) Au₁/COF的TEM图像。(d) Au₁/COF的放大TEM图像。(e) Au₁/COF的像差校正HAADF-STEM图像。(f, g) Au₁/COF的HAADF-STEM图像及对应的C、N、O和Au元素的EDX元素面分布图。
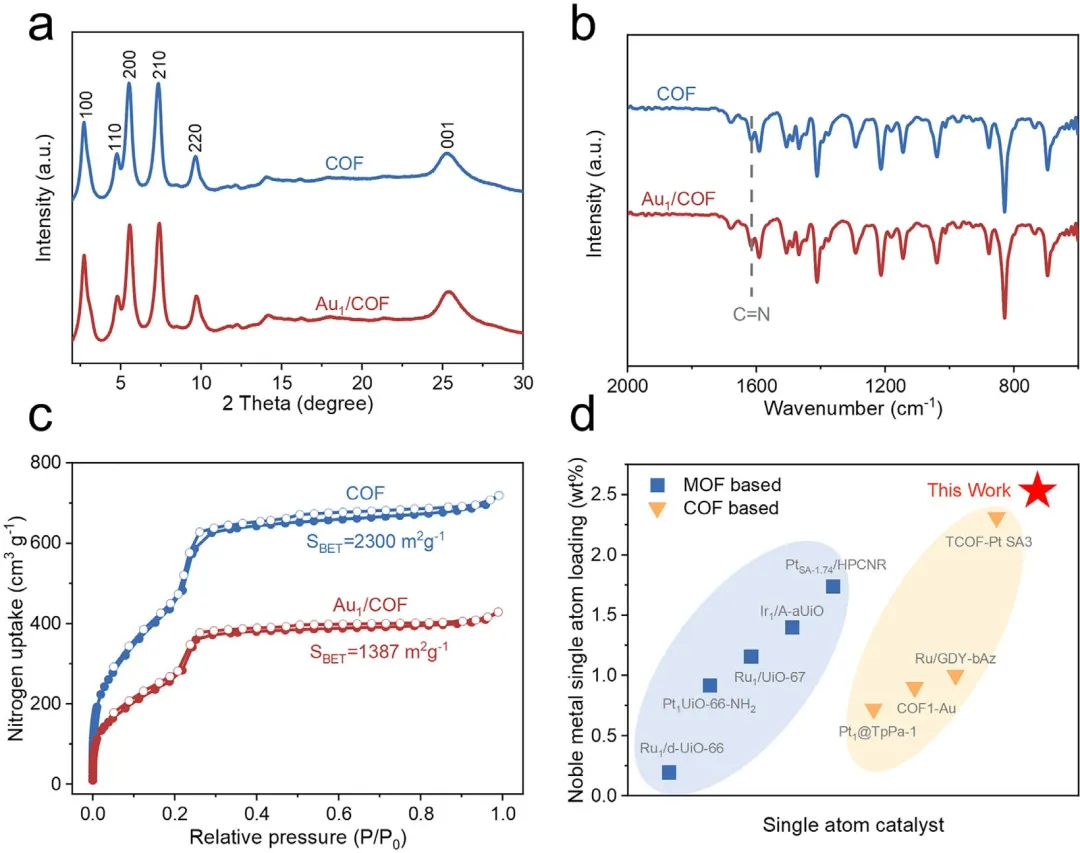
图2 | 单原子负载前后的结构特征及COF负载能力。(a) COF和Au₁/COF的XRD谱图。(b) COF和Au₁/COF的FT-IR光谱。(c) COF和Au₁/COF的氮气吸附-脱附等温线。(d) 与部分已报道基底上单原子负载量的比较。
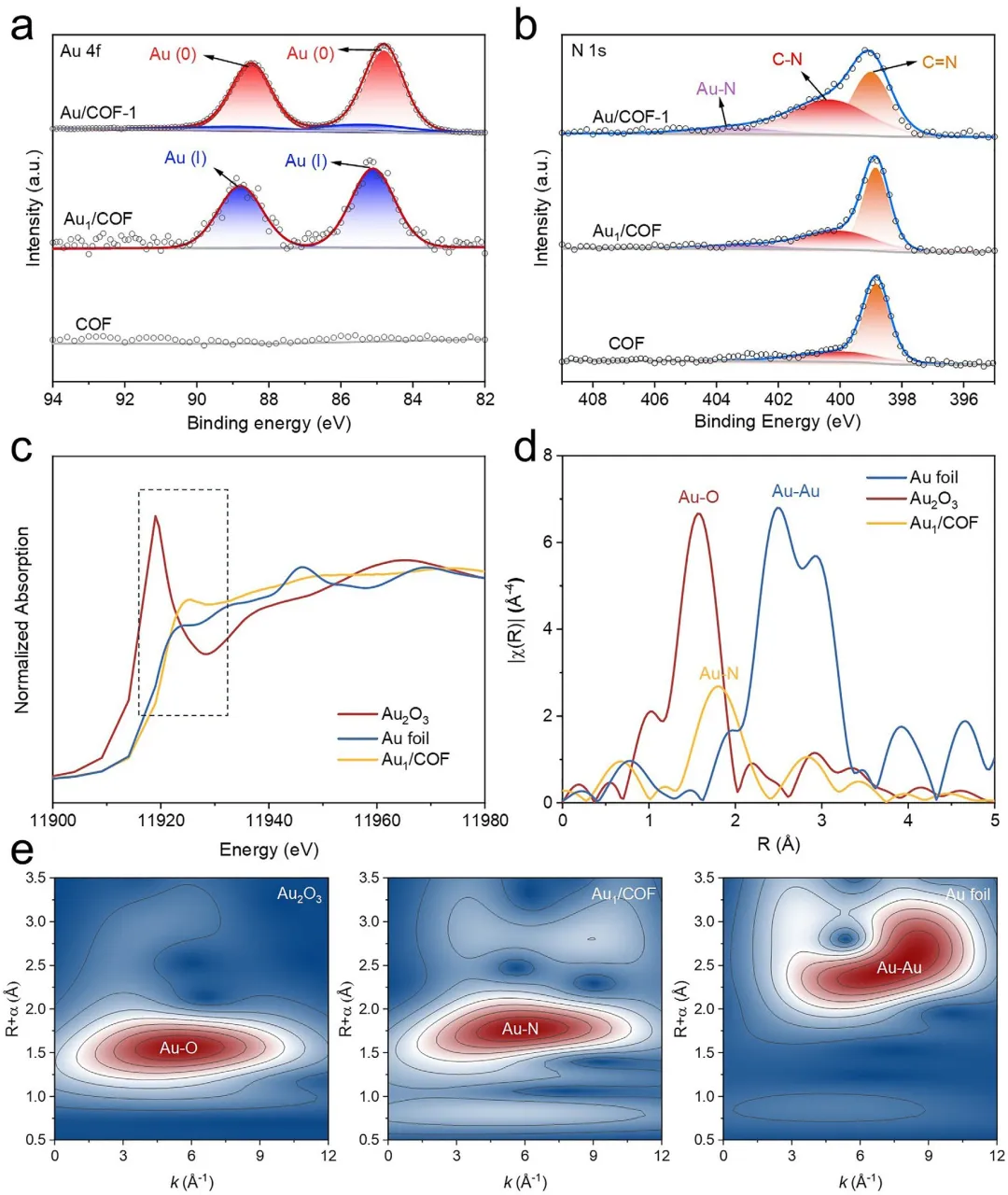
图3 | 配位环境分析。(a) COF、Au/COF和Au/COF-1的Au 4f XPS谱图。(b) COF、Au/COF和Au/COF-1的N 1s XPS谱图。(c) Au/COF及参考样品Au箔和Au₂O₃的Au L-edge XANES谱图。(d) Au/COF及参考样品Au箔和Au₂O₃的k³加权χ-EXAFS数据的傅里叶变换谱图。(e) Au箔、Au₂O₃和Au/COF的EXAFS信号小波变换图。
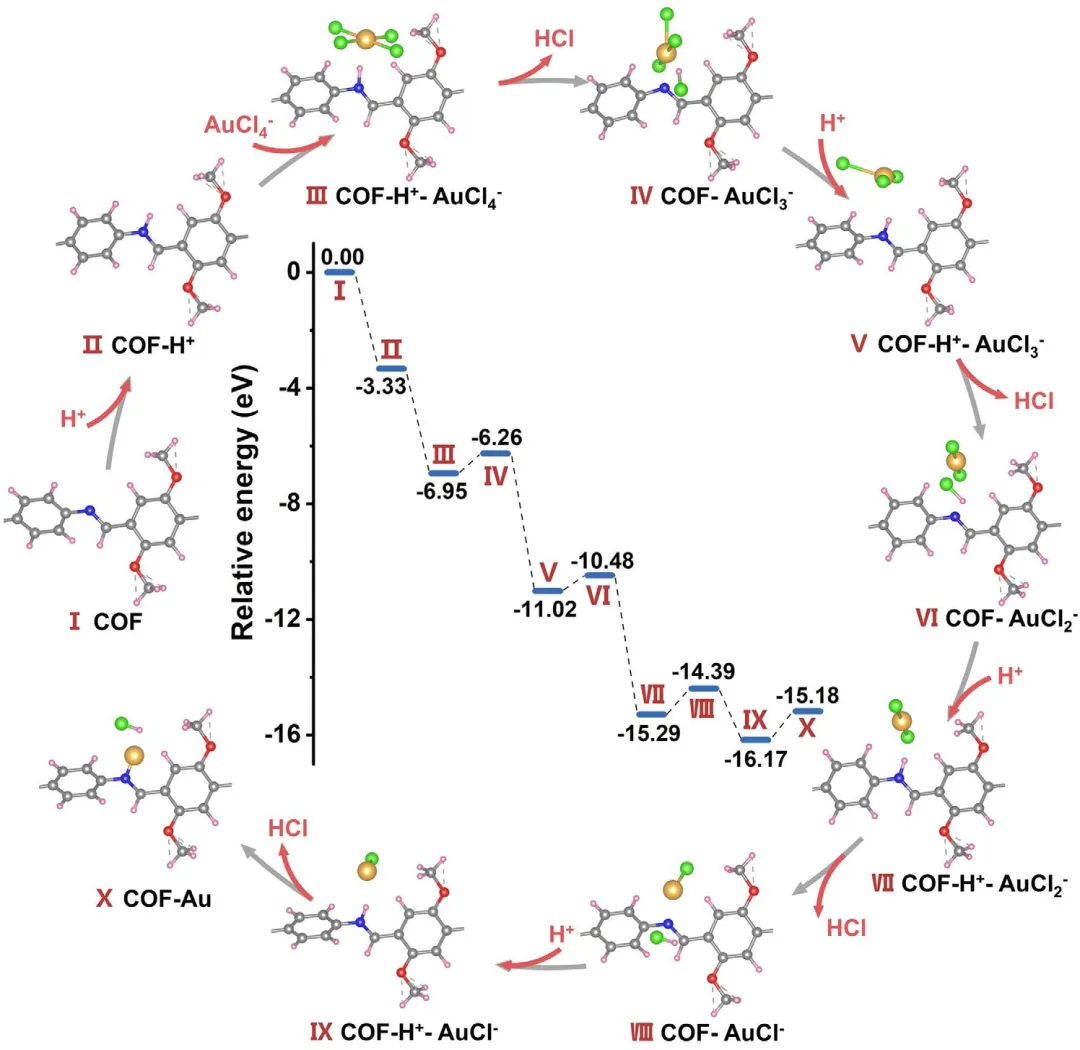
图4 | Au单原子形成的DFT计算。AuCl₄在亚胺键位点上的脱氯能量分布。插图为基于DFT计算的AuCl₄在亚胺键上脱氯的势能分布图。Au,黄色;C,灰色;N,蓝色;O,红色;H,粉色;Cl,绿色。
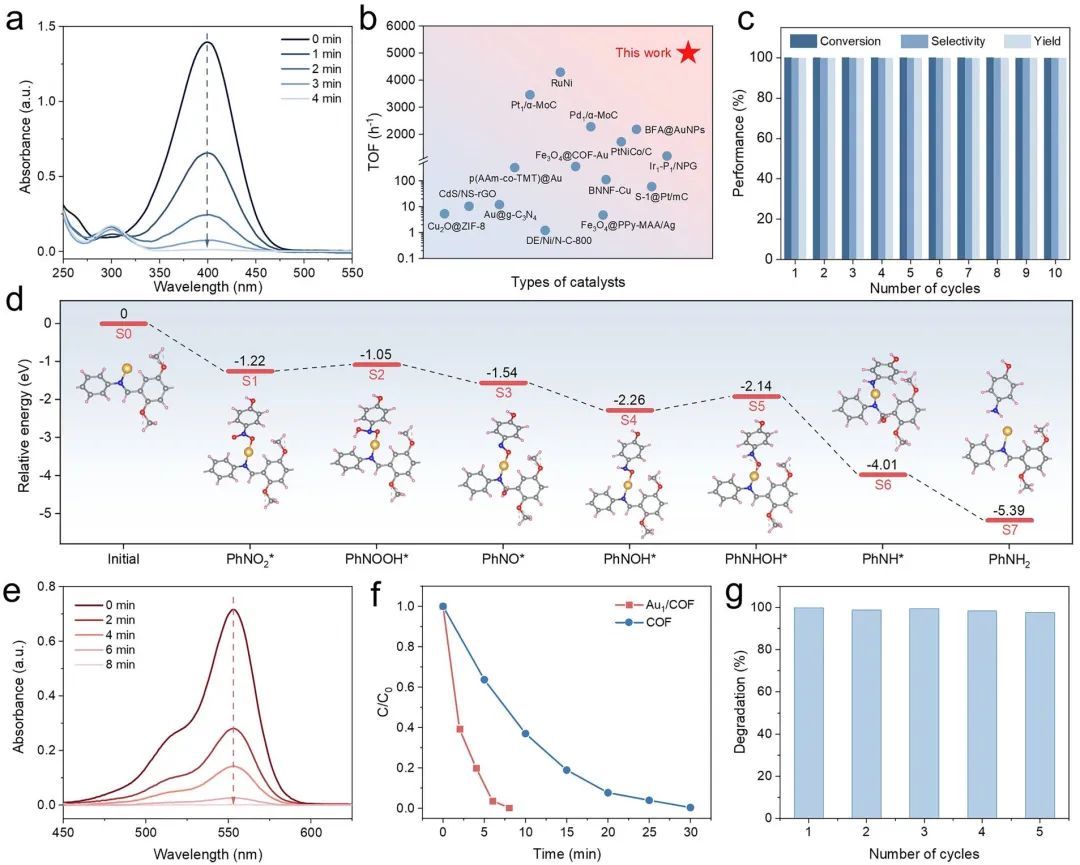
图5 | 催化反应研究。(a) 4-NP的还原反应(5.0 mM 4-NP,3.0 M NaBH₄,0.6 mg催化剂)。(b) 一些已报道催化剂用于4-NP还原的TOF值比较。TOF:每小时每摩尔金反应的对硝基苯酚的摩尔数。(c) Au₁/COF催化还原4-NP的可回收性测试。反应条件为5.0 mM 4-NP,3.0 M NaBH₄,0.6 mg催化剂。(d) Au₁/COF内4-NP加氢生成4-AP反应机理的示意图。Au,黄色;C,灰色;N,蓝色;O,红色;H,粉色。插图为基于DFT计算的Au₁/COF内4-NP加氢的势能分布图。“S0”表示初始状态,“S1–S7”表示一系列吸附态。(e) Au₁/COF在不同照射时间下对RhB降解的UV-vis光谱。(f) COF和Au₁/COF对RhB的降解效率。(g) Au₁/COF在可见光照射下对RhB的循环降解效率。
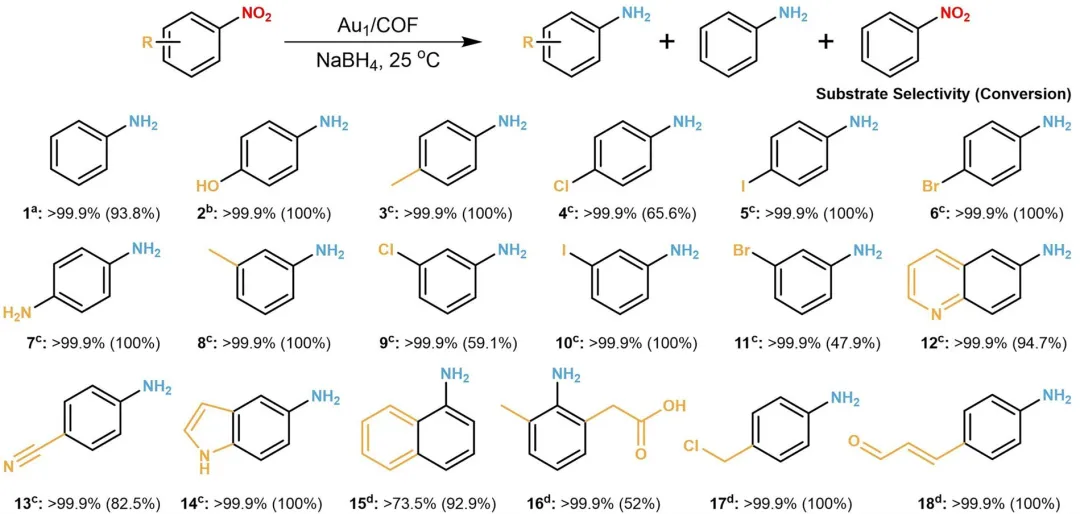
图6 | 在Au₁/COF催化剂存在下硝基芳烃的选择性加氢。星号(a)表示反应条件为:1.0 mM硝基化合物,2.0 M NaBH₄,1.0 mg催化剂,5.0 mL纯化水,室温下连续搅拌3小时。星号(b)表示反应条件为:5.0 mM硝基化合物,3.0 M NaBH₄,0.6 mg催化剂,10 mL纯化水,室温下连续搅拌4分钟。星号(c)表示反应条件为:1.0 mM硝基化合物,2.0 M NaBH₄,1.0 mg催化剂,5.0 mL乙醇,室温下连续搅拌3小时。星号(d)表示反应条件为:1.0 mM硝基化合物,2.0 M NaBH₄,1.0 mg催化剂,5.0 mL乙醇,室温下连续搅拌2小时。
总之,该研究展示了一种利用丙酮溶剂性质合成高负载量Au/COF的直接且高效的方法。丙酮可控的给质子与接受质子特性实现了高Au负载量(2.53 wt.%),超过了众多已报道的COF/MOF负载的贵金属SACs。通过溶剂选择和前驱体浓度精确调控亚胺位点的质子化程度,能够可控形成从单原子到纳米团簇和纳米颗粒的Au物种。Au₁/COF SAC表现出卓越的催化性能,在对硝基苯酚加氢反应中实现了5000 h⁻¹的TOF值,同时在多种硝基芳烃中保持>99.9%的选择性。DFT计算表明,丙酮的弱给质子能力促进了Au前驱体的逐步脱氯,通过COF质子化促进了稳定Au—N键的形成。此外,孤立的Au单原子优化了局域电子环境,这有利于与—NO₂基团反应,并增强了反应动力学和化学选择性。该研究不仅为SACs提供了一种新的合成途径,也强调了溶剂工程在设计用于各种催化应用的COF基催化剂中的重要性。



 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
