1. 发现浆细胞异常富集并驱动不良预后
胶质母细胞瘤中浆细胞显著富集,低体细胞高频突变,与患者更差总生存期直接相关。
2. 揭示CCL2‑CCR2招募浆细胞归巢机制
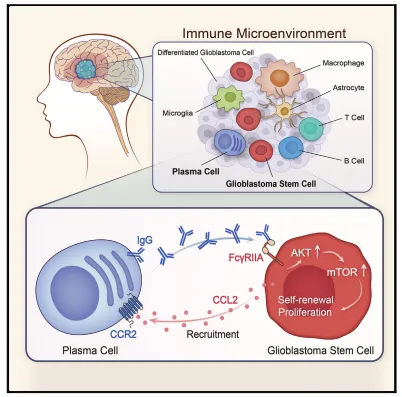
胶质瘤干细胞分泌CCL2,通过CCR2将外周浆细胞招募至干细胞微环境,形成促瘤单元。
3. 阐明IgG‑FcγRIIA‑AKT‑mTOR促瘤轴
浆细胞分泌IgG结合GSC表面FcγRIIA,激活AKT‑mTOR通路,增强干细胞增殖与自我更新。
4. 提出靶向阻断的免疫治疗新策略
阻断FcγRIIA或CCL2‑CCR2可抑制GSC维持、逆转促瘤效应,为GBM提供全新治疗方向。
胶质母细胞瘤恶性度高、预后极差,胶质瘤干细胞驱动进展与耐药,现有治疗效果有限。浆细胞在肿瘤微环境中作用不明,其调控胶质瘤干细胞的机制与干预靶点亟待阐明。研究旨在揭示浸润浆细胞维持胶质瘤干细胞的分子机制,并探索靶向治疗策略。
文章标题:浸润性浆细胞通过IgG-肿瘤结合维持胶质母细胞瘤干细胞
胶质母细胞瘤是一种高度侵袭性的原发性脑肿瘤,其胶质母细胞干细胞(GSCs)主导着肿瘤内部的层级结构。浆细胞(PCs)是B系免疫系统的关键效应细胞,但它们在胶质母细胞瘤中的作用尚未得到充分研究。
研究对胶质母细胞瘤临床样本开展scRNA-seq与scBCR-seq,结合公共数据库分析;构建人源化免疫小鼠模型,通过细胞共培养、移植、分子互作与信号通路实验,探究浆细胞调控胶质瘤干细胞的机制与干预策略。
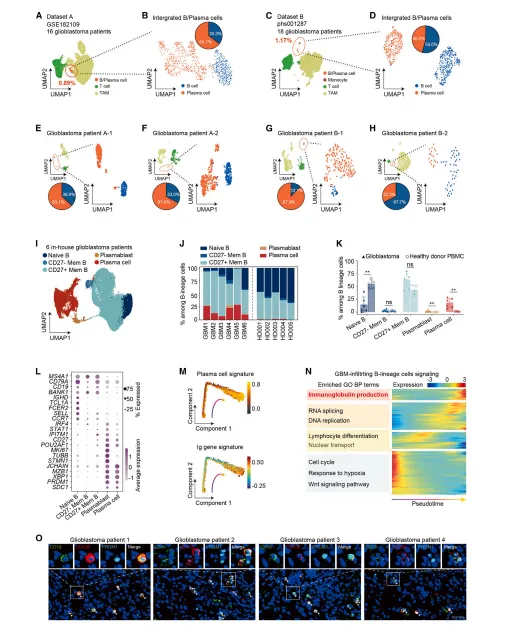
1.在胶质母细胞瘤中,PCs在肿瘤浸润性B系细胞中异常富集
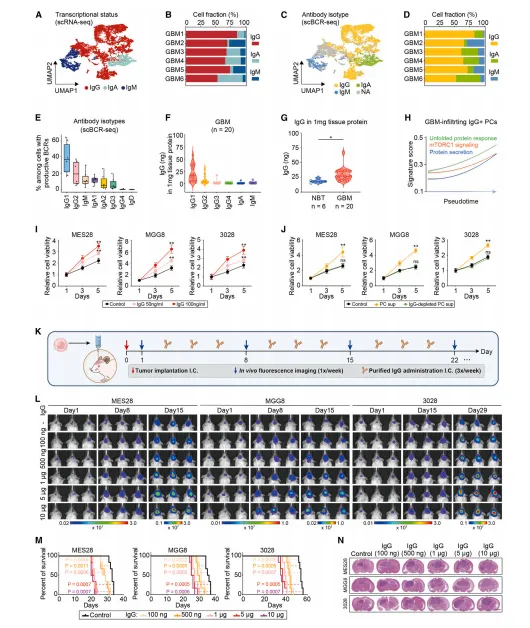
通过scRNA-seq与免疫荧光证实,胶质母细胞瘤中B系细胞占比低,但浆细胞比例显著高于健康人外周血;UMAP分群显示肿瘤内浆细胞特征明显,伴随免疫球蛋白生成通路激活,提示浆细胞在肿瘤微环境中异常富集(图1)。
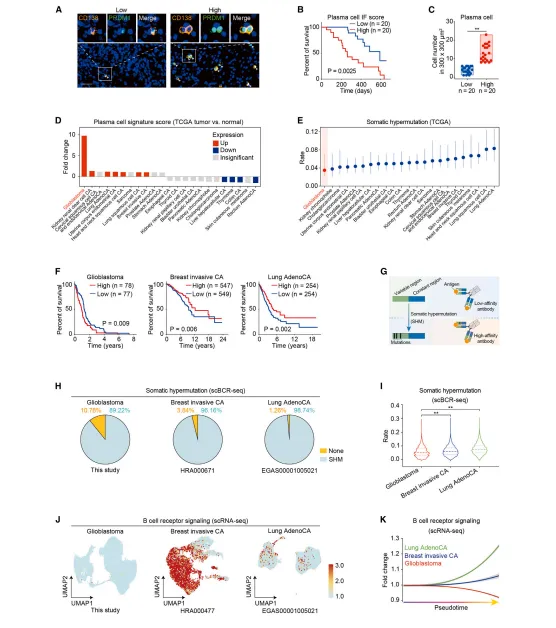
高浆细胞计数患者总生存期显著缩短;跨癌种分析表明,胶质母细胞瘤浆细胞特征评分升高最显著,体细胞高频突变率最低,BCR信号受损,明确浆细胞是胶质母细胞瘤独立不良预后因素(图2)。
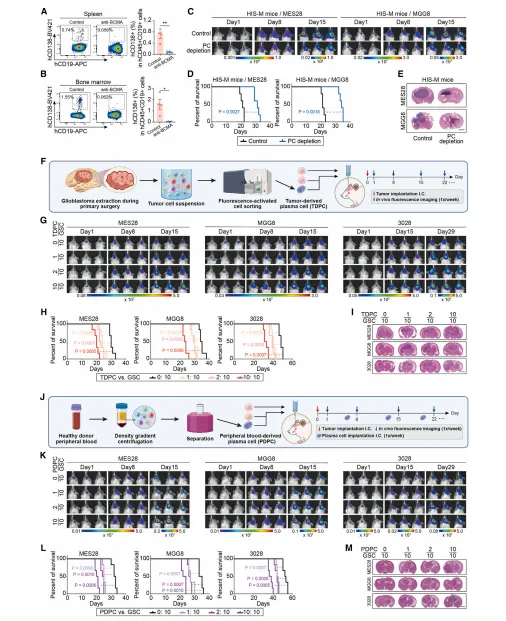
通过人源化小鼠与细胞共培养证实,清除浆细胞可抑制肿瘤生长、延长生存期;浆细胞与胶质瘤干细胞共移植显著加速肿瘤生长、缩短小鼠存活时间,浆细胞对肿瘤进展具有关键驱动作用(图3)。
图3.肿瘤浸润性浆细胞促进胶质母细胞瘤生长并预示不良预后
4.肿瘤浸润浆细胞分泌的IgG支持胶质母细胞瘤的增殖
肿瘤内浆细胞主要分泌IgG,IgG浓度显著高于正常脑组织;IgG可特异性结合胶质瘤干细胞并促进其增殖,清除IgG后促增殖作用消失,IgG是浆细胞促瘤的关键效应分子(图4)。
图4.肿瘤浸润浆细胞分泌的gG促进胶质母细胞瘤增殖
5.IgG-FcgRIIA- AKT -mTOR信号轴参与维持生殖干细胞(GSC)功能,并促进肿瘤浸润浆细胞(PCs)刺激的肿瘤发生
鉴定出FcγRIIA为胶质瘤干细胞特异性高表达受体,IgG结合后激活SYK-PI3K-AKT-mTOR通路;敲低FCGR2A可阻断IgG与浆细胞上清的促增殖作用,揭示该信号轴为核心促瘤机制(图5)。
图5.IgG-FcgRIIA- AKT -mTOR信号通路参与了由肿瘤浸润浆细胞(PCs)刺激的胃鳞癌(GSC)肿瘤生长过程
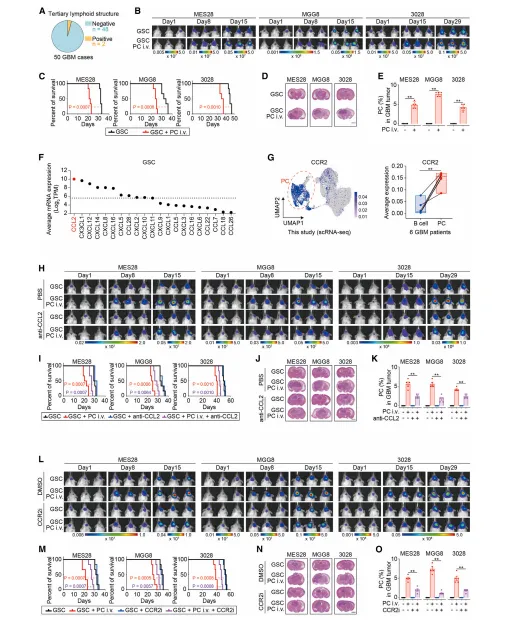
6.胶质母细胞瘤浸润性浆细胞通过CCL2-CCR2趋化因子程序被募集至生殖干细胞(GSC)微环境
胶质瘤干细胞高分泌CCL2,浆细胞高表达CCR2;CCL2-CCR2通路介导浆细胞定向迁移至肿瘤干细胞龛,阻断该通路可减少浆细胞浸润、抑制肿瘤生长,明确浆细胞的招募机制(图6)。
图6.PC细胞通过CCL2-CCR2趋化因子程序被募集至GSC生态位
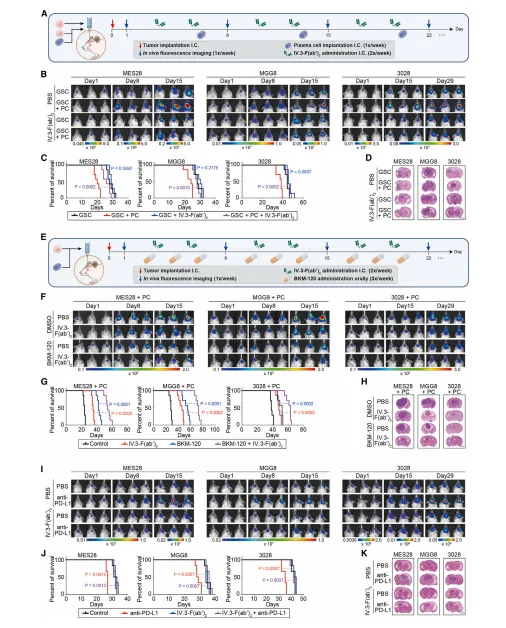
7.IgG受体阻断可抑制肿瘤浸润性PC对GSC的刺激作用,并减少胶质母细胞瘤的生长
FcγRIIA单抗可阻断IgG促瘤信号,联合PI3K抑制剂协同抑瘤;同时可逆转IgG型免疫检查点抑制剂的促瘤效应,显著延长荷瘤小鼠生存期,证实该靶点具有临床转化价值(图7)。
图7.IgG受体阻断可抑制肿瘤浸润性PC-GSC的刺激作用,并在体内抑制GSC的肿瘤发生
研究构建了胶质母细胞瘤中B系细胞的图谱,并为联合靶向肿瘤细胞内在特性与微环境依赖性提供了理论框架。