南京林业大学陈振ACS Catal.:光氧化还原助力镍催化实现芳基溴代的自由基羰基化反应
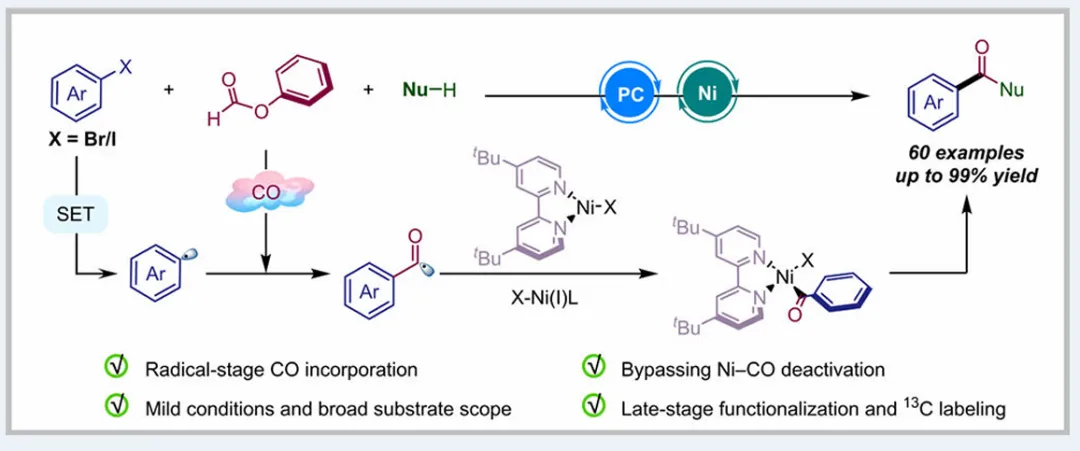
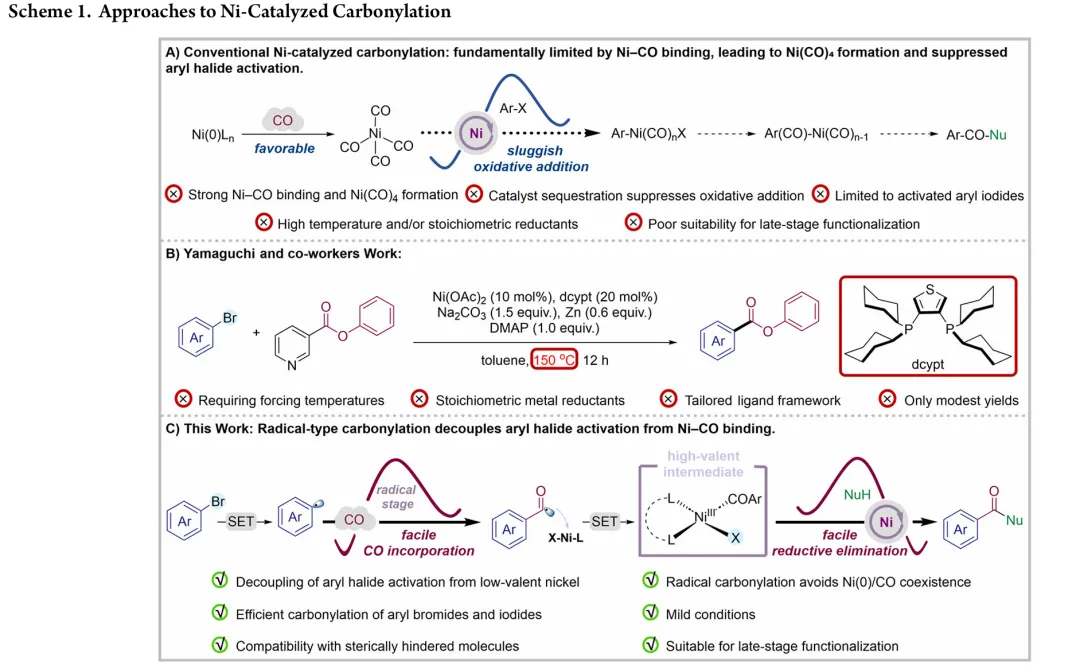
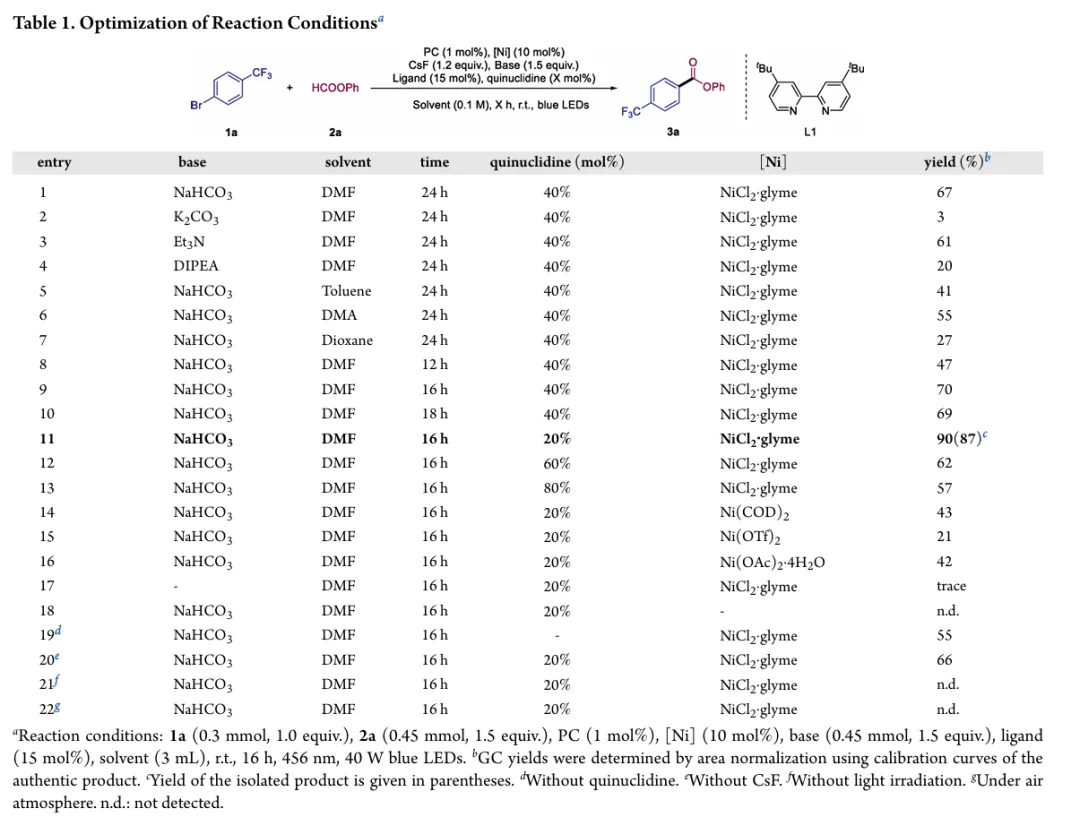
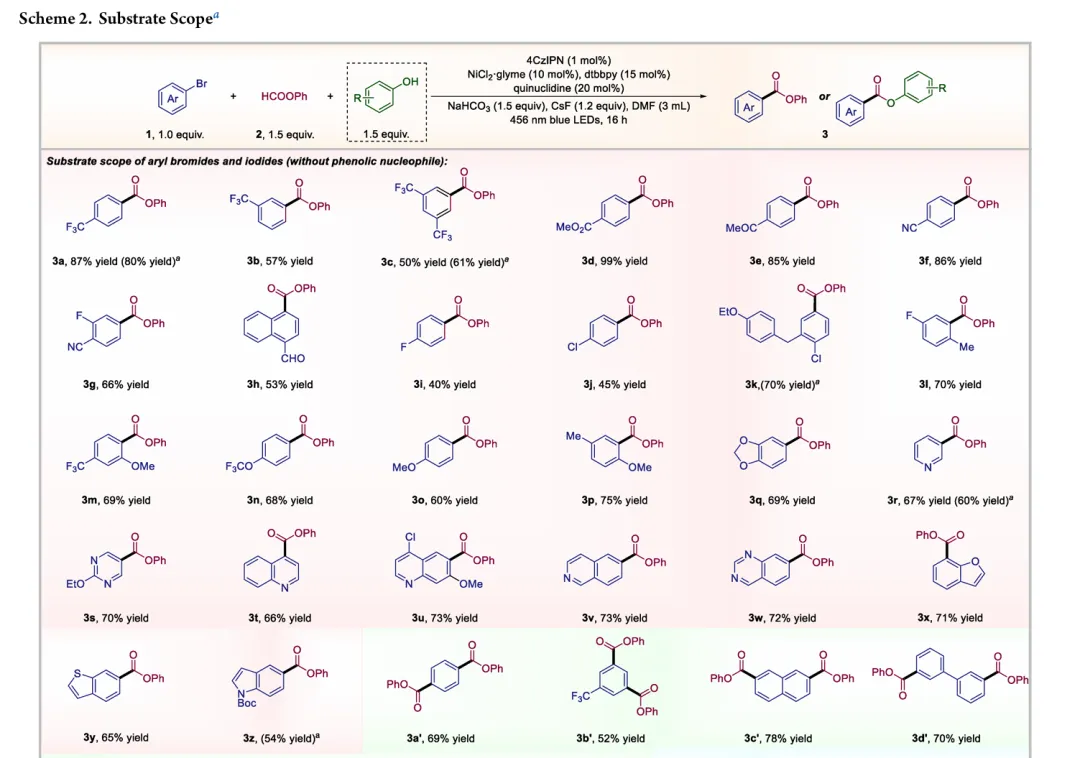
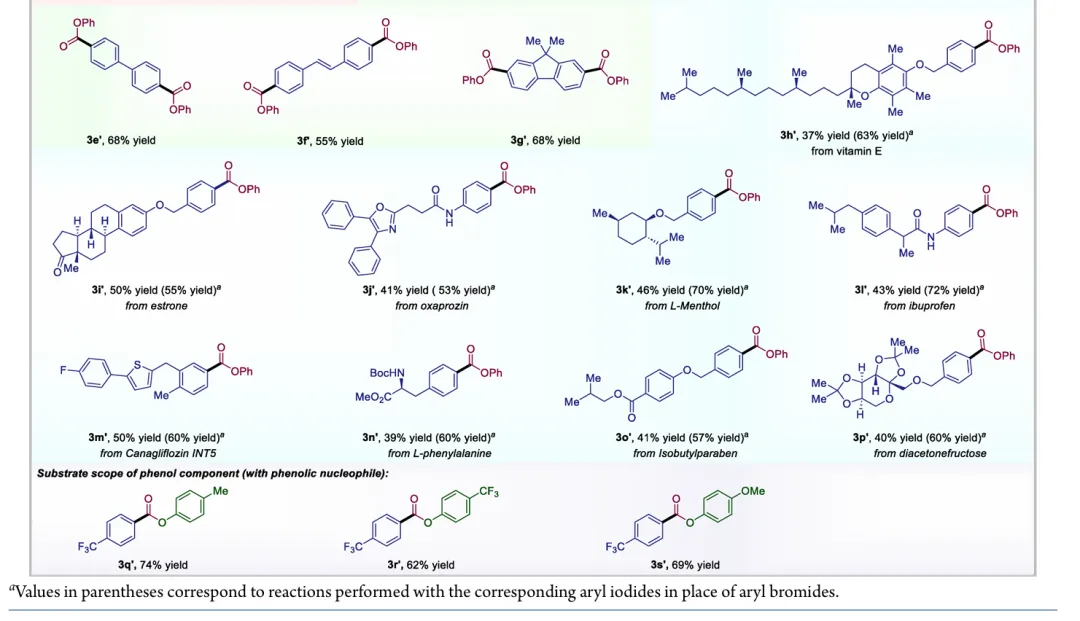
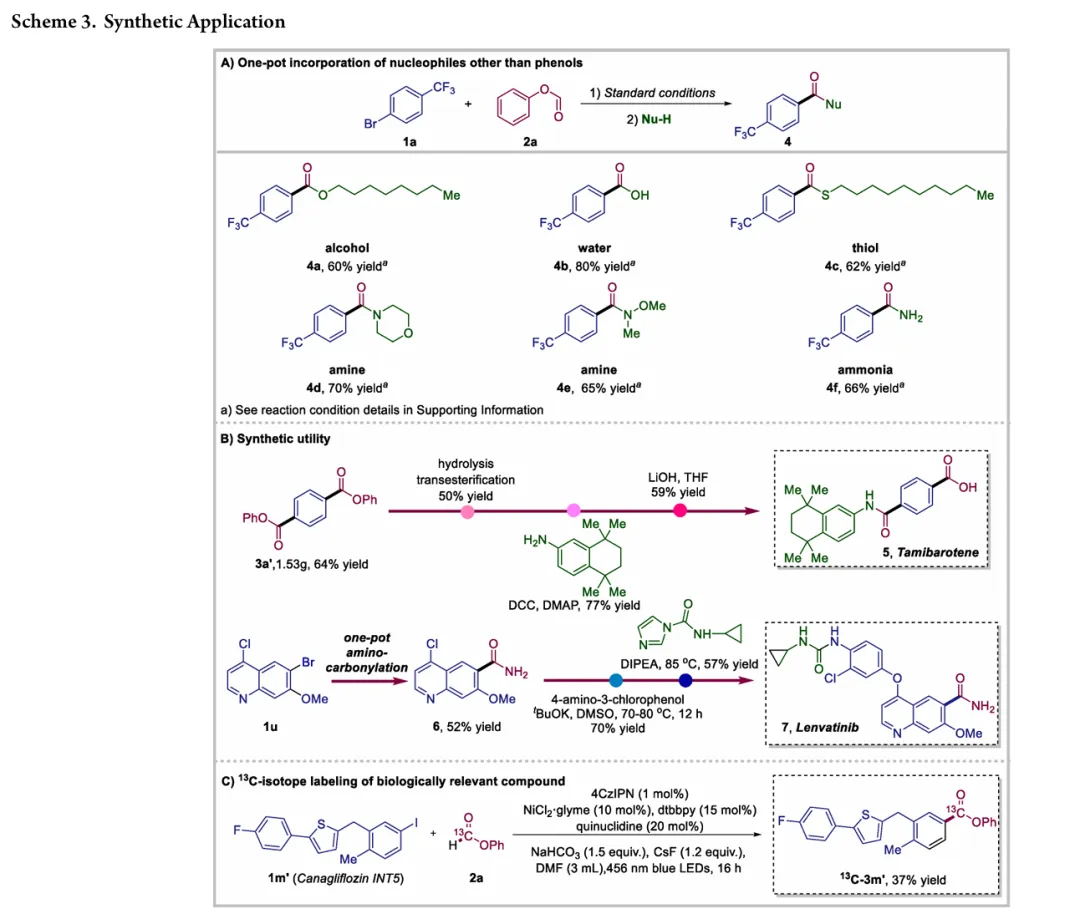
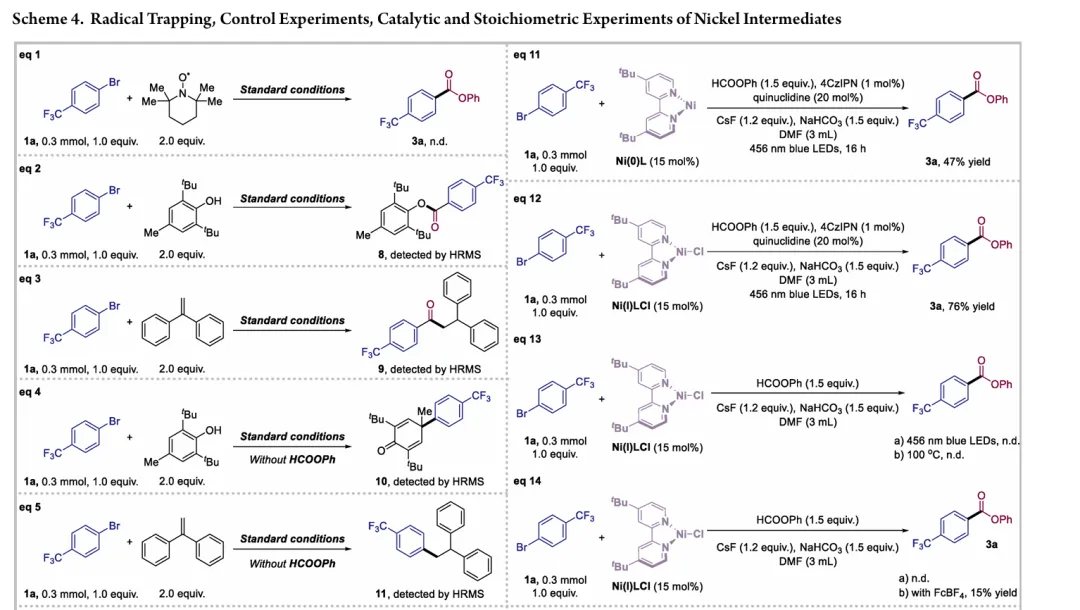
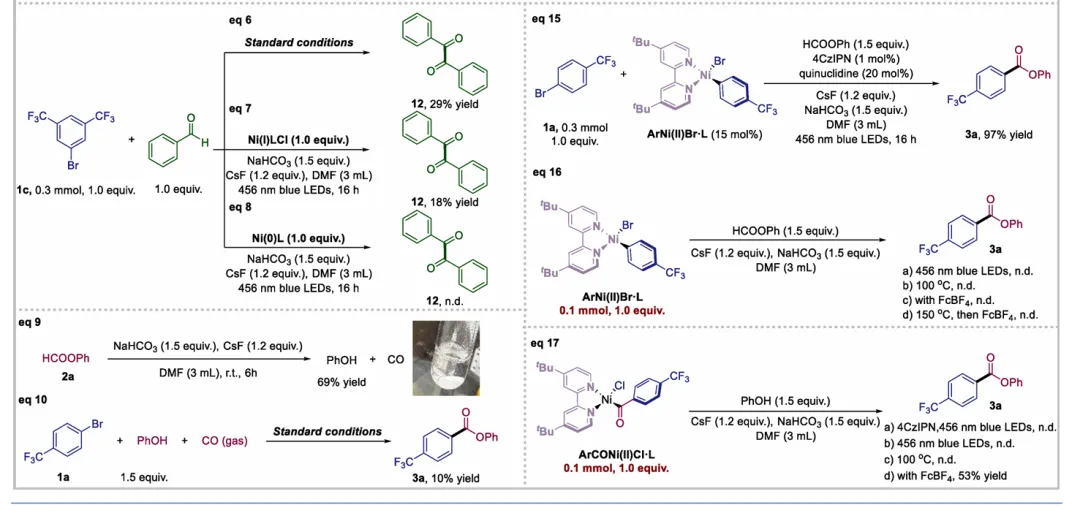
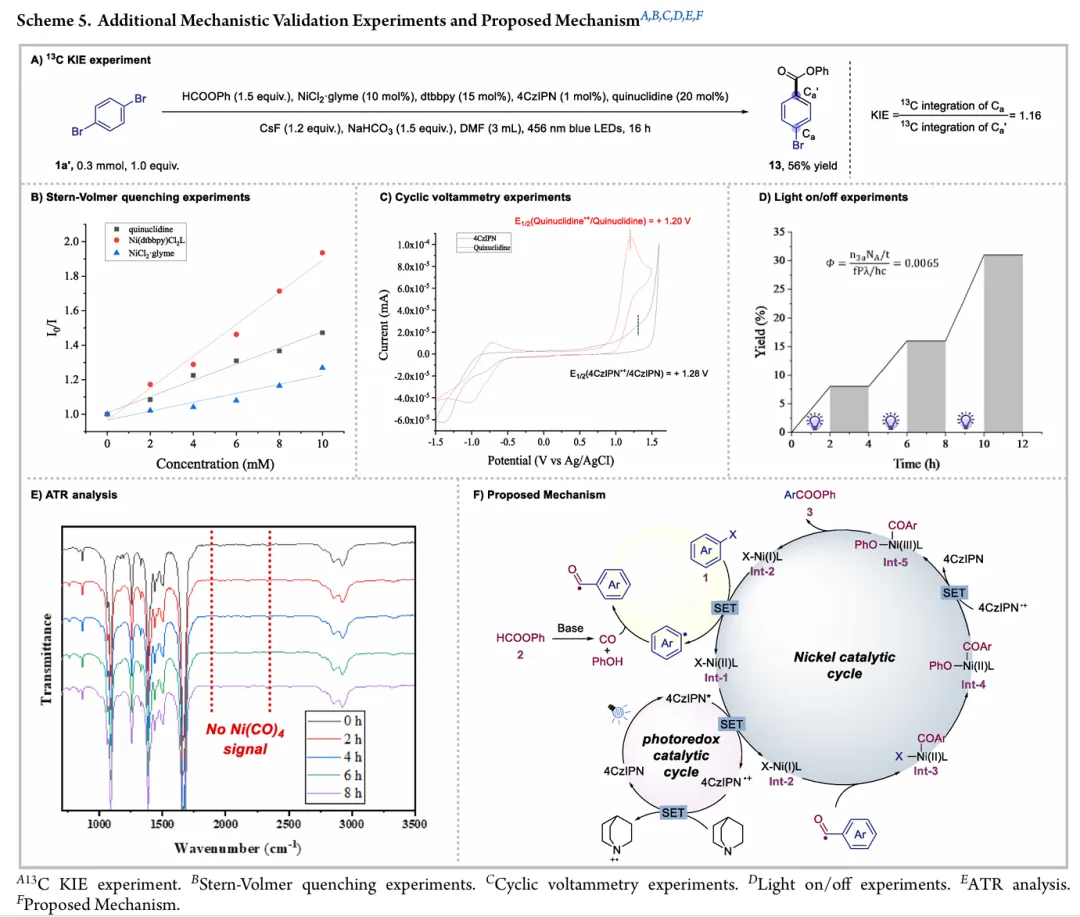
图1. TOC (图片来源于ACS Catal.)Nickel-catalyzed carbonylation represents an attractive alternative to palladium-based methods but has long been constrained by the strong affinity of low-valent nickel for carbon monoxide, leading to formation of catalytically inactive Ni(CO)4 species. As a result, mild nickel-catalyzed carbonylation of aryl bromides has remained largely inaccessible. Here, we report a photoredox-enabled nickel-catalyzed carbonylative esterification of aryl bromides using phenyl formate as a practical and safe in situ CO surrogate. By decoupling aryl bromide activation from nickel–CO binding and relocating CO incorporation to a radical pathway, this strategy enables efficient carbonylation of aryl bromides under ambient conditions. The method exhibits broad substrate scope, high functional-group tolerance, and good compatibility with sterically hindered and complex molecular architectures. Mechanistic studies reveal a pathway involving aryl radical generation, radical carbonylation, nickel capture, and oxidation-enabled C(O)–O bond formation. This work establishes a conceptually distinct strategy to nickel-catalyzed carbonylation that bypasses the intrinsic limitations of conventional Ni–CO chemistry.图2. 研究背景及本策略 (图片来源于ACS Catal.)图3. 条件优化 (图片来源于ACS Catal.)图4. 底物拓展 (图片来源于ACS Catal.)图5. 合成应用 (图片来源于ACS Catal.)图6. 自由基捕获、控制实验、镍中间体的催化和当量实验 (图片来源于ACS Catal.)图7. 其他机理研究及可能的催化循环 (图片来源于ACS Catal.)In conclusion, we have developed a mild and practical photoredox-enabled nickel-catalyzed platform for the carbonylative esterification of aryl bromides using phenyl formate as a safe, in situ CO surrogate. Operating under ambient conditions, this protocol exhibits broad substrate scope, high functional-group tolerance, and pronounced insensitivity to steric hindrance, enabling efficient late-stage modification of structurally complex molecules. The synthetic utility of the method is further demonstrated through the concise preparation of pharmaceutical intermediates and site-specific 13C isotope labeling. Comprehensive mechanistic investigations, including radical trapping experiments, kinetic isotope effect measurements, and photophysical analyses, support a pathway initiated by photoinduced electron transfer and proceeding through a nickel catalytic cycle involving both low- and high-valent intermediates. By decoupling aryl halide activation from nickel–CO binding and relocating carbonyl incorporation to a radical manifold, this work establishes a general and conceptually distinct strategy for carbonylative C–O bond formation that circumvents the intrinsic limitations of conventional nickel–CO chemistry. We anticipate that this paradigm will provide a broadly applicable foundation for the development of new nickel-catalyzed carbonylation reactions relevant to medicinal chemistry, isotope labeling, and functional materials.

 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
