南京大学 Appl. Catal. B Environ. Energy | 单原子与邻近团簇协同优化过一硫酸盐活化的非自由基热力学路径以高效脱毒抗生素废水
我们建立了催化交流群,老师和同学可以添加微信进入。微信号:
nyhhjch001
1. 研究背景及意义
抗生素废水的大量排放加剧了耐药基因的传播并破坏了水体生态系统,严重威胁全球水资源安全。传统的基于过一硫酸盐的高级氧化工艺虽然能有效降解有机污染物,但严重依赖高性能催化剂以维持高活性的氧化物种供应。碳基单原子催化剂因其极高的原子利用率而备受关注,但其单原子位点中带负电的氮配体往往会排斥过一硫酸盐中的氧原子,导致反应物吸附较弱、表面氧化剂富集不足。若单纯通过增加金属负载量来增强吸附,极易导致金属在热解中团聚成纳米颗粒,这不仅降低了原子利用率,还会因吸附过强导致产物难以脱附,进而引发活性位点中毒。
为了打破这种单原子与纳米颗粒在吸附与脱附之间的权衡局限,本研究提出了一种结合单原子高利用率与团簇多金属构型优势的创新设计思路。作者利用富氮的低成本金属有机框架作为前驱体,通过精准调节孔道内的钴负载量,在多孔碳基底上成功构筑了空间邻近的钴单原子与亚纳米团簇耦合位点。

该研究不仅为突破传统单原子催化剂在高级氧化过程中的吸附-脱附活性瓶颈提供了全新的原子级位点协同策略,还深入揭示了非自由基表面碰撞氧化的微观机制,对于推动催化氧化技术在复杂工业废水深度处理及生态脱毒中的规模化应用具有重要的指导意义。
2. 主要研究结果
2.1 实验方法
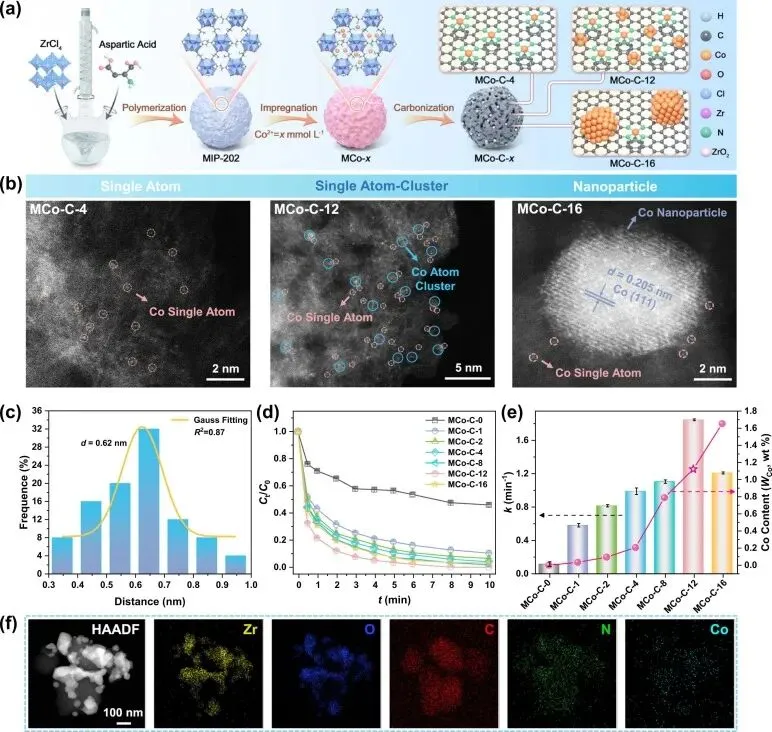
作者采用具有超小孔径的绿色金属有机框架MIP-202作为前驱体,通过浸渍-热解策略制备了负载钴的孔碳催化剂。与传统缺乏配体或孔隙约束的载体体系相比,MIP-202的受限微孔不仅能通过富氮位点锚定孤立的单原子,还能促使吸附在同一微孔中的多个金属离子在高温碳化时相互靠近形成金属键。通过控制初始钴离子浓度,实现了催化位点从孤立钴单原子向单原子-团簇耦合结构,再到较大尺寸钴纳米颗粒的可控形态演变。
 2.2 研究亮点
2.2 研究亮点
球差校正高角环形暗场扫描透射电子显微镜测试直接证实,在具有中等钴负载量的催化剂中实现了钴单原子与钴团簇的共存,两者之间的平均空间间距仅为0.62纳米。X射线吸收精细结构光谱进一步证明了体系中钴的平均化合价约为正二价,且配位壳层中同时存在平均配位数为5.0的钴-氮键以及配位数为2.9的钴-钴键。这些关键表征确凿地证明了单原子与四面体亚纳米团簇的相邻耦合结构已在碳骨架中被成功构筑。
 2.3 核心性能表现
2.3 核心性能表现
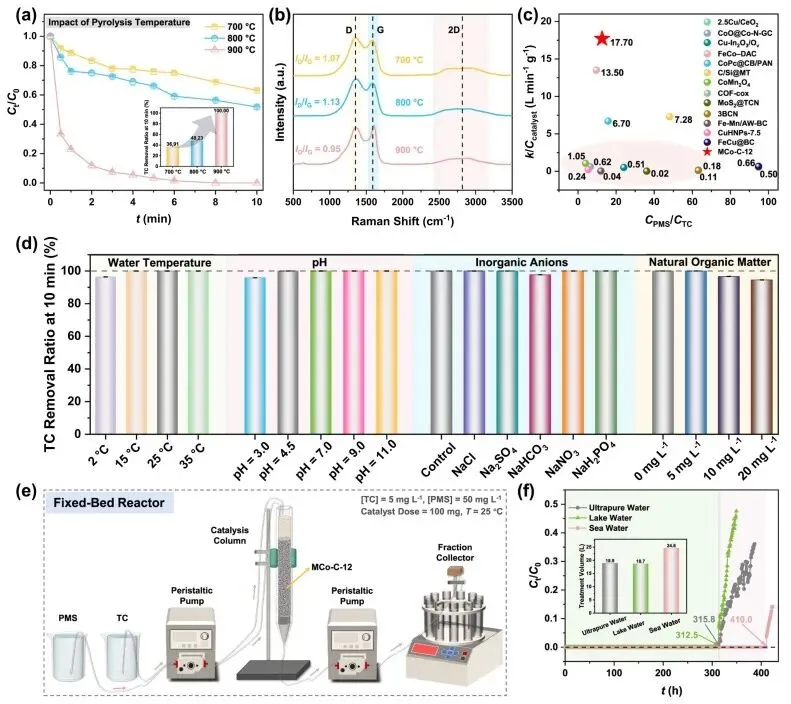
在活化过一硫酸盐降解四环素的测试中,具有单原子-团簇耦合位点的催化剂在10分钟内实现了100%的去除率,其催化剂剂量归一化的反应速率常数高达17.70 L min-1 g-1,比大多数文献报道的基准值高出1至2个数量级。该体系在2至35摄氏度以及pH值为3.0至11.0的宽泛范围内,即使面对共存的无机阴离子和天然有机质,依然保持了极高的催化活性。在连续流固定床反应器中,仅需100毫克催化剂即可在真实的湖水和海水中分别实现长达312.5小时和410.0小时的四环素连续完全去除,总处理水量分别达到18.7升和24.6升。
 2.4 机理验证
2.4 机理验证
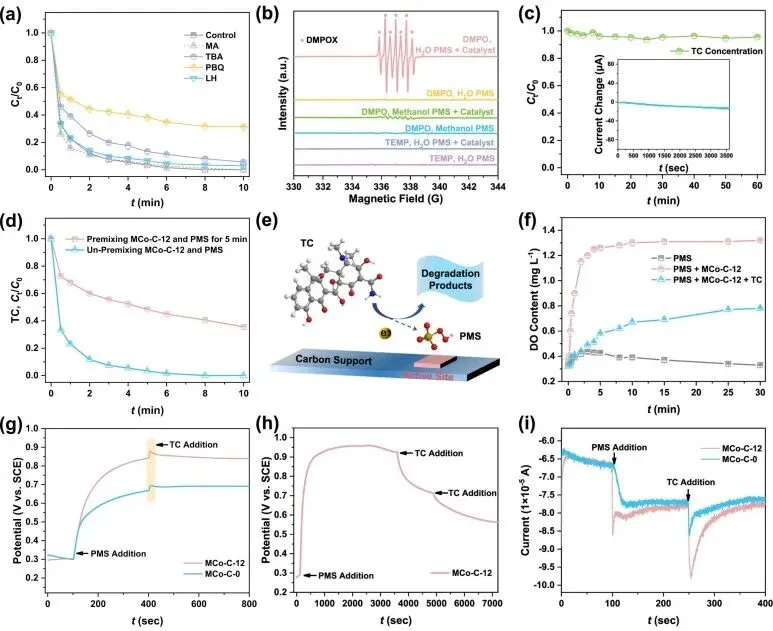
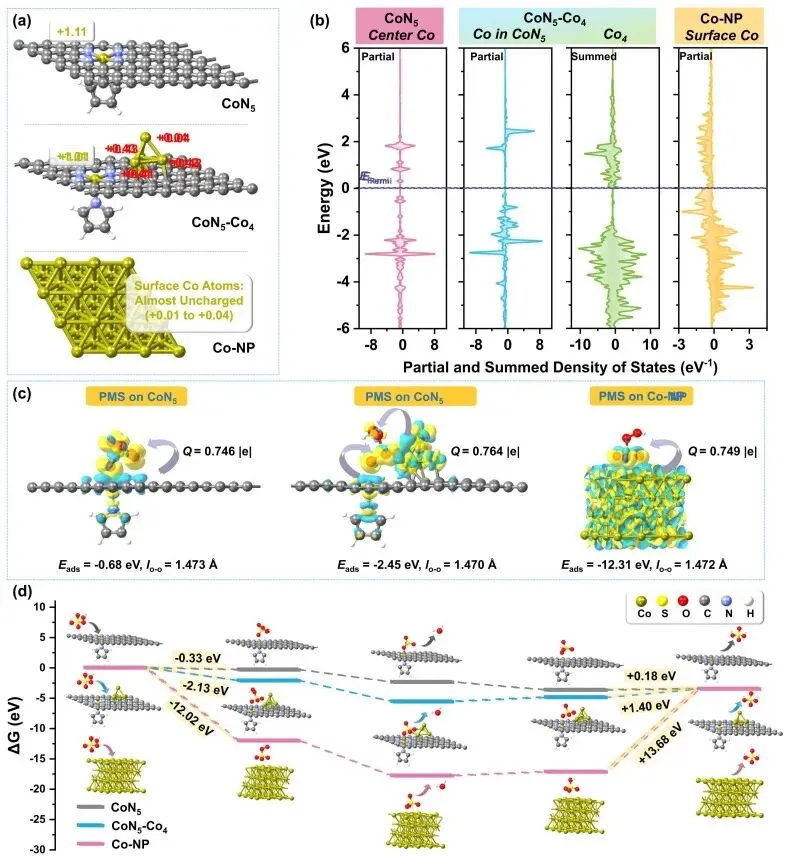
通过自由基猝灭实验与电子顺磁共振测试,作者排除了羟基自由基、硫酸根自由基、超氧自由基和单线态氧在降解过程中的主导作用。结合原位拉曼光谱、溶解氧监测与电化学分析证实,反应遵循表面碰撞氧化路径。催化剂活化过一硫酸盐生成了高电位的表面结合态复合物,该复合物无需释放自由基,而是直接通过与周围四环素分子的有效碰撞提取电子以引发降解。理论计算表明,单原子-团簇耦合位点向过一硫酸盐转移了0.764 e的电子,其吸附能为-2.45 eV,既克服了单原子位点吸附较弱的劣势,又避免了纳米颗粒位点因硫酸氢根产物脱附能垒过高(+13.68 eV)而导致的失活,从而在热力学上赋予了体系最高的内禀催化活性。
 3. 研究结论
3. 研究结论
本研究通过微孔限域策略成功构筑了空间邻近的钴单原子与亚纳米团簇耦合催化位点,有效打破了常规碳基单原子催化剂在高级氧化过程中的吸附-脱附活性限制。这项工作不仅提出并验证了依赖高电位表面结合物直接提取电子的非自由基表面碰撞氧化新机制,还实现了将剧毒的抗生素分子高效转化为可供微生物直接利用的低毒碳源,为后续通过原子级位点协同设计开发适用于复杂水体净化的绿色催化材料提供了重要的科学启示。
https://doi.org/10.1016/j.apcatb.2026.126907
我们建立了催化交流群,感兴趣的老师和同学可以添加微信,邀请您进群。



