南京理工大学朱俊武/熊攀&南科大叶财超,最新Nature子刊!高熵RP钙钛矿氧化物自旋态调控助力高效海水电解!刘一凡/郝礼彬一作
「上海岱算科技有限公司」已向境内外230余家高等院校/科研院所提供了累计1400多项模拟计算服务,赋能科学研究提速增效!合作实验课题组在线发表学术论文期刊有ACS系列、AM系列、Angew、CEJ、EST、JACS、Matter、Nature子刊等,助力科研工作锦上添花!高熵氧化物已成为多种催化领域(包括海水电解)中一类颇具前景的候选材料,然而由于缺乏合理的理解与指导,为高熵体系设计多元素组成以及阐明不同元素在催化中的协同效应仍然面临重大挑战。
2026年05月09日,南京理工大学朱俊武、熊攀团队在Nature Communications期刊发表题为“Spin-state regulation of high-entropy Ruddlesden-Popper perovskite oxides for efficient seawater electrolysis”的研究论文,团队成员刘一凡、郝礼彬为论文共同第一作者,朱俊武、熊攀、南方科技大学叶财超为论文共同通讯作者。
第一作者:刘一凡、郝礼彬
通讯作者:朱俊武、熊攀、叶财超
通讯单位:南京理工大学、南方科技大学
论文DOI:10.1038/s41467-026-72948-x
该研究报道了一种高熵Ruddlesden-Popper钙钛矿氧化物的自旋态调控,用于高效海水电解。具有Jahn-Teller效应的Cu²⁺和Mn³⁺诱导了Fe³⁺/Co³⁺自旋态从低自旋向高自旋构型的转变。高自旋Fe³⁺/Co³⁺有利于海水氧化过程中OH⁻吸附和去质子化。此外,吸附在高自旋活性位点上的氧中间体能够排斥Cl⁻离子。因此,(La₀.₇₆Sr₀.₂₄)₃(Fe₀.₂₂Co₀.₂₁Ni₀.₁₈Cu₀.₁₇Mn₀.₂₂)₂O₇在200mA cm⁻²下实现了超过1200小时的稳定海水氧化,表现出极具竞争力的性能。以(La₀.₇₆Sr₀.₂₄)₃(Fe₀.₂₂Co₀.₂₁Ni₀.₁₈Cu₀.₁₇Mn₀.₂₂)₂O₇为阳极、Pt/C为阴极组装的阴离子交换膜电解槽能够在1.76V电压下维持1A cm⁻²的海水分解性能,并连续运行800小时。这种自旋调控策略对其他高熵钙钛矿氧化物和尖晶石氧化物具有普适性,为设计高效高熵催化剂提供了一种有前景的途径。
绿氢通过利用可再生电力的水电解制得,因其无污染特性和优异的质量能量密度而变得日益重要。与使用高纯水作为原料的传统水电解相比,直接海水电解是一种更具吸引力的替代方案。这是由于世界许多地区淡水资源的稀缺以及地球上海水储量巨大。然而,海水复杂的组分对设计高效电催化剂提出了重大挑战。电催化剂面临的主要挑战是阳极上的Cl⁻吸附和析氯反应(ClER)。研究人员已采取多种方法来工程化常规金属氧化物电催化剂,包括构建Cl⁻排斥层、调控局部环境以及引入析氧反应(OER)保护层。然而,具有简单组成和有限活性位点的常规金属氧化物可能难以在复杂海水环境中实现高效电催化,从而限制了它们满足未来海水氧化先进要求的潜力。
高熵催化剂,例如高熵氧化物(HEOs),近年来发展迅速,在催化应用中备受关注。由于固有的鸡尾酒效应和缓慢扩散效应,HEOs相较于常规金属氧化物表现出更优异的电催化活性和增强的本征稳定性。一些实例表明,具有五种组分的HEOs是熵诱导性能增强的阈值。例如,五组分岩盐相HEOs在电化学活性和循环稳定性方面比任何四组分类似物都表现出显著的提升。高熵(CoFeNiMnW)₃O₄氧化物利用其高熵组成来优化平衡不同的析氧路径,克服了传统的活性-稳定性权衡,并提供了比三元对应物更低的过电位。此外,富缺陷高熵尖晶石氧化物在淡水和海水环境中均实现了比四元对应物更高的水氧化活性。含贵金属的HEOs可以利用其多金属配位环境向Ru/Ir活性位点提供电子。这有效抑制了贵金属的过氧化和溶解,从而显著增强了催化剂的稳定性。尽管近期研究已利用鸡尾酒效应和熵稳定化来提升性能,但由于元素组成复杂以及缺乏合理的组成设计原则,HEOs催化剂的发展仍然受到催化机制不明确的阻碍。因此,将五种组分随机掺入HEOs中是否能有效增强其电催化活性和稳定性?在纯水分解体系中,通过大量合成和性能测试来筛选HEOs中的元素是合适的。然而,在复杂的海水分解体系中,依靠实验筛选来确定HEOs的最佳元素组成是低效的,特别是要同时实现优异的海水氧化活性和耐氯腐蚀性。例如,有报道称 (FeCoNiMnAl)₃O₄ 高熵电催化剂因其对阴离子(OH⁻和Cl⁻)的强吸附能力而表现出优于中熵对应物的海水氧化活性。然而,对Cl⁻的强吸附会腐蚀阳极电催化剂,并不可避免地导致晶体结构的不稳定。因此,有必要为复杂催化体系中HEOs的合理设计和组分筛选建立理论指导。
自旋催化概念已被提出用于调控具有相对简单组成的材料中的自旋电子构型。例如,Xue等人将配位不饱和边缘位点引入CoOOH结构中,诱导Co³⁺从低自旋到高自旋构型的自旋转变,导致电子从面对面的t2g轨道转移到顶对顶的eg轨道,从而实现快速OER催化动力学。由此推测,自旋构型可能是HEOs中连接高熵效应与催化性能的一个潜在决定性因素。原则上,HEOs中具有不同自旋态的M3d轨道(M = 金属位点)分别与氧物种和氯物种的O2p和Cl3p轨道表现出不同的重叠和相互作用。因此,调控自旋态可以显著优化OH⁻和Cl⁻的化学吸附,从而增强海水氧化过程中的氧选择性和氯离子排斥能力。然而,HEOs复杂的原子排列对识别活性位点分布和控制其自旋态提出了巨大挑战。因此,HEOs中自旋态与海水电解之间的关系尚未见报道。
在此,该研究报道了一种用于高效海水电解的高熵Ruddlesden-Popper相钙钛矿氧化物(RP-FeCoNiCuMn)的自旋态调控。首先选择Fe、Co和Ni元素用于高熵催化剂设计,因为它们被广泛认为是水氧化反应的高活性位点。考虑到具有Jahn-Teller效应的Cu²⁺和Mn³⁺可以优化活性位点的局部配位几何构型,进一步将其掺入以诱导活性位点从低自旋(LS)到高自旋(HS)构型的自旋转变。RP-FeCoNiCuMn中电解活性Fe³⁺和Co³⁺的高自旋态有利于海水氧化反应过程中OH⁻离子的吸附和去质子化过程。同时,阳极上优先吸附的氧中间体将通过静电排斥排斥Cl⁻离子,从而增强在碱性海水中的催化稳定性。因此,具有高自旋活性位点的调控后的RP-FeCoNiCuMn在碱性海水电解中表现出稳健的稳定性和高效的活性,优于高熵对应物以及其他已报道的氧化物基催化剂。以RP-FeCoNiCuMn为阳极、商业Pt/C为阴极的海水阴离子交换膜(AEM)电解槽实现了高活性(1A cm⁻²下1.76V)和良好稳定性(1A cm⁻²下超过800小时)。此外,自旋态调控策略具有普适性,可扩展至其他高熵钙钛矿和尖晶石相,用于显著提升催化活性。
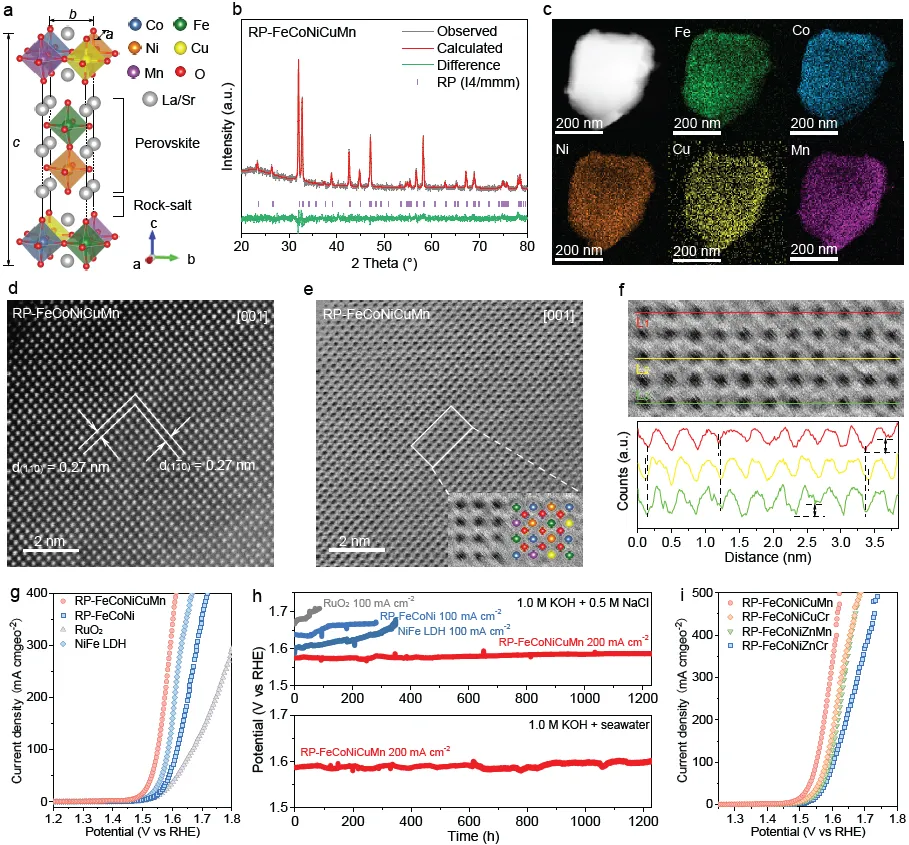
图1. 结构表征与电催化测量。(a) 高熵Ruddlesden-Popper钙钛矿氧化物结构的示意图。(b) RP-FeCoNiCuMn的精修XRD谱图。(c) RP-FeCoNiCuMn的STEM图像及相应的元素面分布图。(d) RP-FeCoNiCuMn沿[001]晶带轴的HAADF-STEM和(e) ABF-STEM图像。(f) RP-FeCoNiCuMn的放大ABF-STEM图像及相应的线剖面图,显示出明显的强度变化和原子列的错位。(g) RP-FeCoNiCuMn、RP-FeCoNi、商业RuO₂和NiFe LDH在模拟碱性海水中的极化曲线。(h) 在模拟碱性海水和碱性海水中的长期稳定性测试。(i) RP-FeCoNiCuMn、RP-FeCoNiCuCr、RP-FeCoNiZnMn和RP-FeCoNiZnCr在模拟碱性海水中的极化曲线。
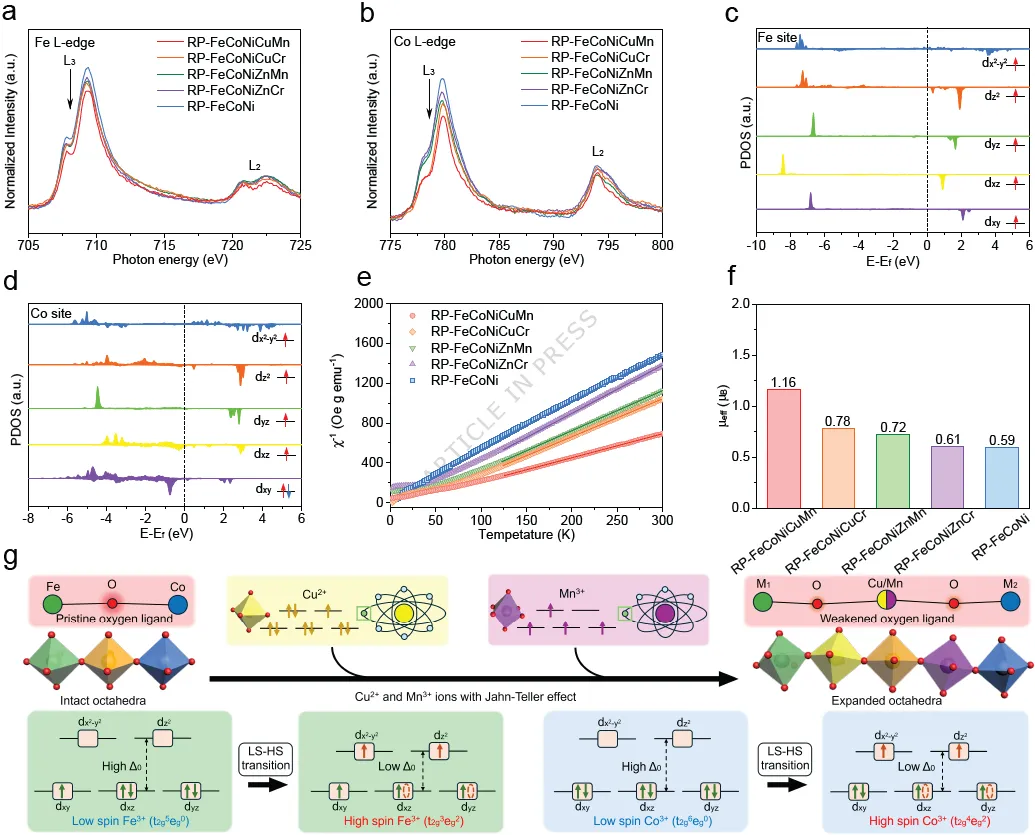
图2. 所有研究的高熵和中熵RP相氧化物的自旋态结构分析。(a) Fe和(b) Co L边XANES谱图,样品为RP-FeCoNiCuMn、RP-FeCoNiCuCr、RP-FeCoNiZnMn、RP-FeCoNiZnCr和RP-FeCoNi。(c) RP-FeCoNiCuMn中Fe和(d) Co的3d轨道,按自旋向上和自旋向下区分。(e) RP-FeCoNiCuMn、RP-FeCoNiCuCr、RP-FeCoNiZnMn、RP-FeCoNiZnCr和RP-FeCoNi样品的磁化率温度依赖性倒数。(f) 样品的有效磁矩。(g) 引入具有J-T效应的Cu²⁺/Mn³⁺后,Co³⁺/Fe³⁺发生低自旋到高自旋转变的示意图。
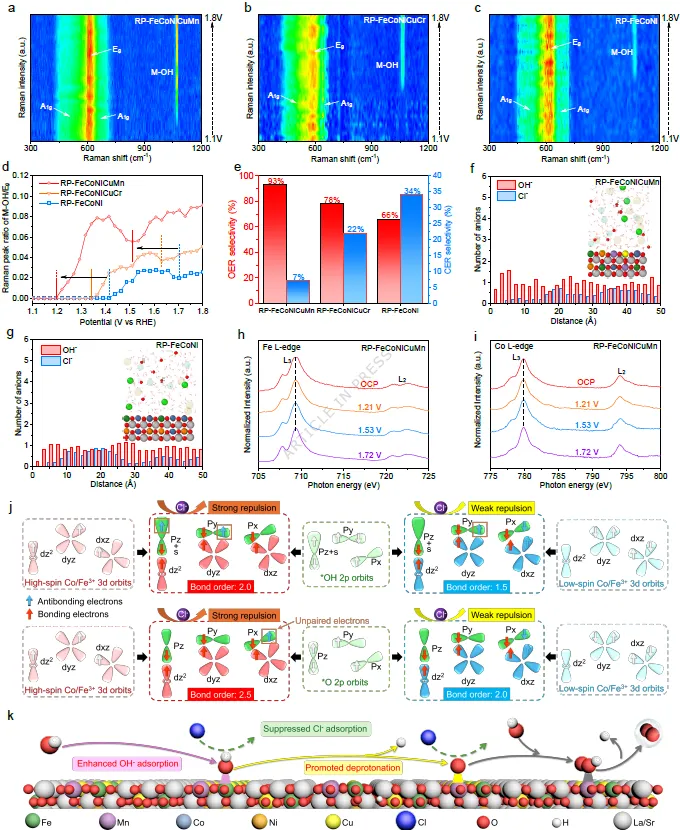
图3. 海水氧化活性及稳定性与自旋态的相关性。RP-FeCoNiCuMn (a)、RP-FeCoNiCuCr (b) 和 RP-FeCoNi (c) 在OER过程中,电压范围为1.1至1.8 V vs. RHE下的原位拉曼光谱。(d) RP-FeCoNiCuMn、RP-FeCoNiCuCr和RP-FeCoNi的 M-OH/Eg 拉曼峰面积比的变化图。(e) 在模拟海水条件下,使用旋转环盘电极技术评估RP-FeCoNiCuMn、RP-FeCoNiCuCr和RP-FeCoNi催化剂的OER和CER选择性。(f) RP-FeCoNiCuMn和(g) RP-FeCoNi的暴露表面来自分子动力学模拟结果。(f)和(g)中的插图分别显示了RP-FeCoNiCuMn和RP-FeCoNi表面上方的经典分子动力学模拟快照。红色、浅绿色、白色、蓝色、绿色、橙色、黄色、紫色、半透明黄色和半透明蓝色球体分别代表O、Cl、H、Co、Fe、Ni、Cu、Mn、K和Na原子。(h) RP-FeCoNiCuMn的(h) Fe和(i) Co L边非原位XAS谱图。(j) 不同自旋态Co³⁺/Fe³⁺与氧中间体之间的轨道相互作用及其对Cl⁻离子的排斥能力。(k) RP-FeCoNiCuMn中高自旋活性位点在碱性海水中促进析氧反应并抑制Cl⁻吸附的示意图。
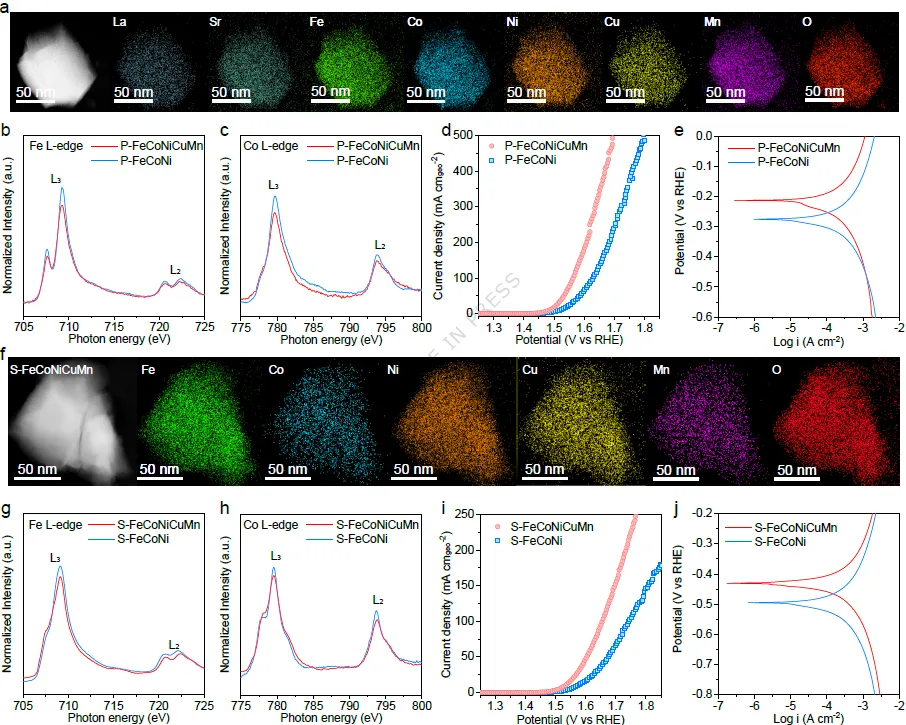
图4. 高熵催化剂中自旋态调控的普适性研究。(a) P-FeCoNiCuMn的STEM图像及相应的元素面分布图。(b) P-FeCoNiCuMn和P-FeCoNi的Fe L边和(c) Co L边XANES谱图。(d) P-FeCoNiCuMn和P-FeCoNi的极化曲线和(e) 腐蚀极化曲线。(f) S-FeCoNiCuMn的STEM图像及相应的元素面分布图。(g) S-FeCoNiCuMn和S-FeCoNi的Fe L边和(h) Co L边XANES谱图。(i) S-FeCoNiCuMn和S-FeCoNi的极化曲线和(j) 腐蚀极化曲线。
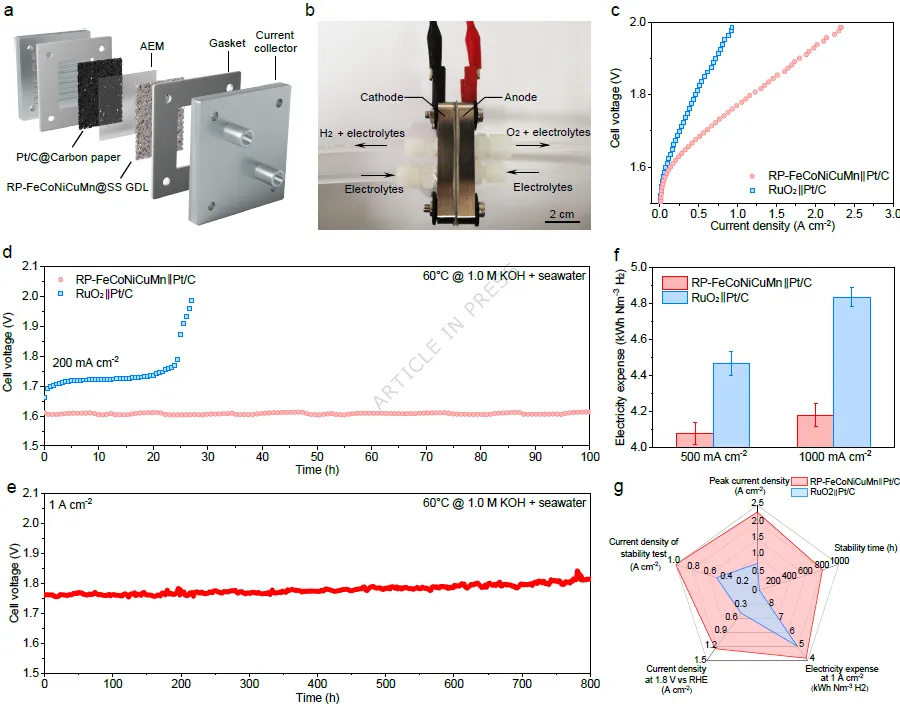
图5. AEM海水电解槽的全解水性能。(a) AEM海水电解槽的示意图和(b) 实物照片。(c) 使用RP-FeCoNiCuMn和商业RuO₂(1.0 mg cm⁻²)作为阳极催化剂、商业Pt/C(0.5 mgPt cm⁻²)作为阴极催化剂,在60°C下测得的AEM海水电解槽的极化曲线。(d) RP-FeCoNiCuMn||Pt/C和RuO₂||Pt/C电解槽在200 mA cm⁻²和60°C下运行的计时电位曲线。(e) RP-FeCoNiCuMn||Pt/C电解槽在1 A cm⁻²和60°C下运行的计时电位曲线。(f) 根据极化曲线计算得到的AEM海水电解槽电耗比较。误差线表示三次重复测量数据的标准差。(g) RP-FeCoNiCuMn||Pt/C和RuO₂||Pt/C电解槽电催化性能的雷达图比较。
总之,该研究报道了高熵Ruddlesden-Popper(RP)相钙钛矿氧化物(RP-FeCoNiCuMn)的自旋态调控,用于高效且持久的海水氧化。具有J-T效应的Cu²⁺和Mn³⁺可以诱导Co³⁺/Fe³⁺位点的自旋态从低自旋转变为高自旋,从而促进海水氧化反应过程中的OH⁻吸附和去质子化。高自旋Co³⁺/Fe³⁺活性位点上优先吸附的氧中间体能够排斥碱性海水中的Cl⁻离子,从而提高稳定性。因此,所合成的RP-FeCoNiCuMn在100 mA cm⁻²下的过电位仅为320 mV,并能在200 mA cm⁻²下稳定运行超过1200小时而无明显性能衰减,优于高熵对应物、代表性的碱性海水催化剂NiFe LDH以及其他一系列已报道的氧化物基催化剂。当与Pt/C阴极配对组装成AEM电解槽时,RP-FeCoNiCuMn阳极能够在1.76 V下维持1 A cm⁻²的海水分解活性长达800小时。该自旋态调控方法适用于其他代表性的高熵钙钛矿氧化物甚至高熵尖晶石氧化物体系。该研究发现提供了一种自旋工程策略,用于制备兼具高活性和高稳定性的高熵电催化剂。



 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
