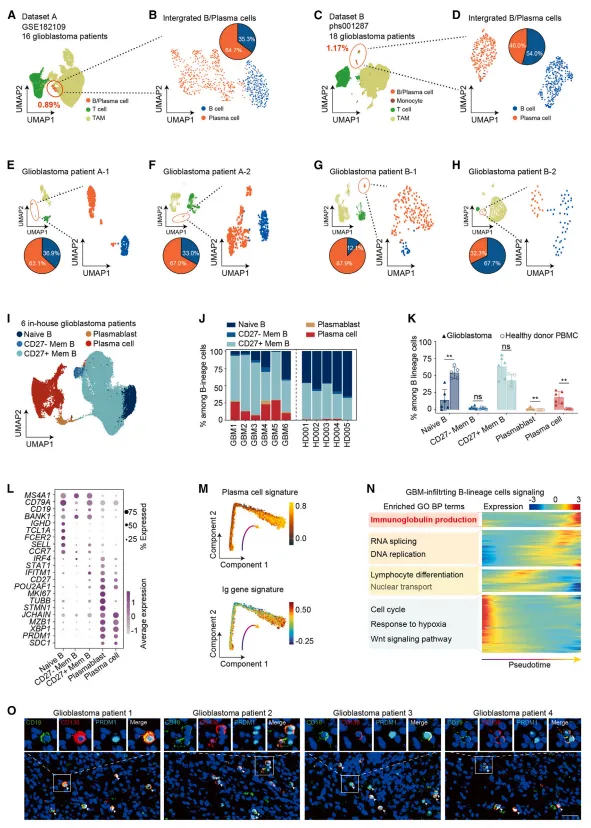
图1:单细胞转录组和BCR测序揭示,浆细胞在胶质母细胞瘤中异常富集,且其丰度与患者不良预后相关
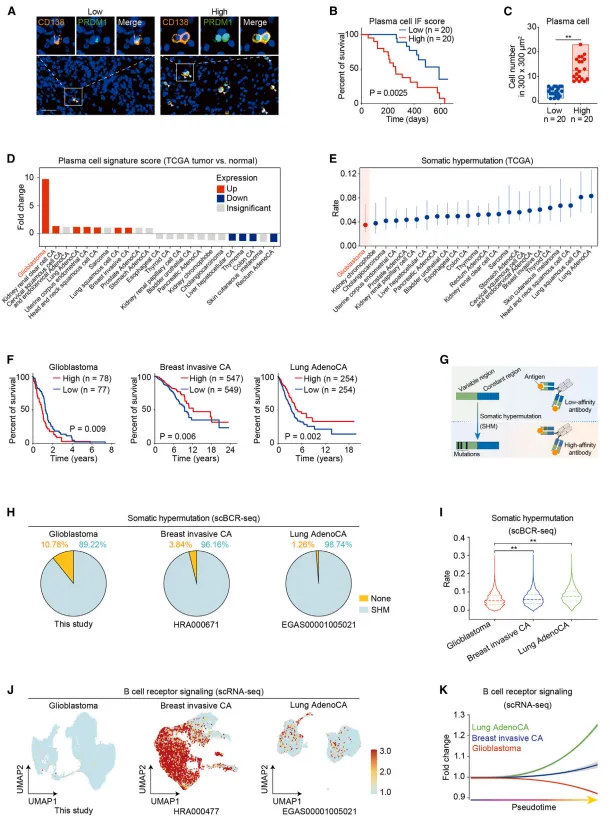
与其他癌种(如肺癌、乳腺癌)相比,胶质母细胞瘤中的浆细胞虽然发生了类别转换(以IgG为主),但体细胞高频突变水平显著偏低,提示这些浆细胞可能不依赖经典的生发中心依赖成熟路径【图2】。
💡单细胞BCR分析不仅要看亚型和克隆,更要计算突变负荷,这能帮你推测细胞的来源和活化历史。
图2:与其他癌种相比,胶质母细胞瘤浸润浆细胞表现出显著降低的体细胞高频突变水平。
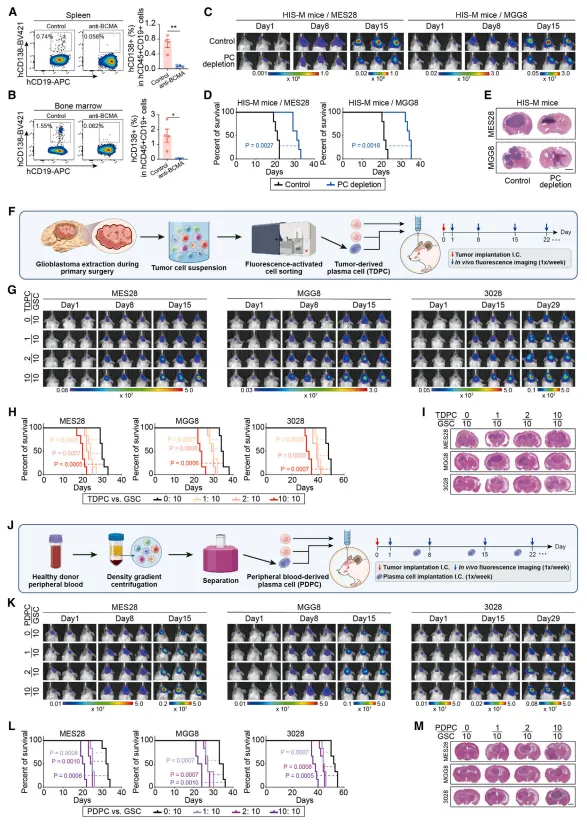
将浆细胞与胶质瘤干细胞共培养后,胶质瘤干细胞的增殖能力和成球能力明显增强【图3B-E】。
小鼠颅内共移植实验中,浆细胞与胶质瘤干细胞共同注射会加速肿瘤生长并显著缩短生存期,且效应与浆细胞数量呈正比【图3F-M】。
💡体内外功能实验要联动验证,尤其是颅内共移植模型,能最直接证明细胞间的促癌“对话”
图3:体内外功能实验显示,浆细胞直接促进胶质瘤干细胞的增殖、自我更新及体内成瘤能力。
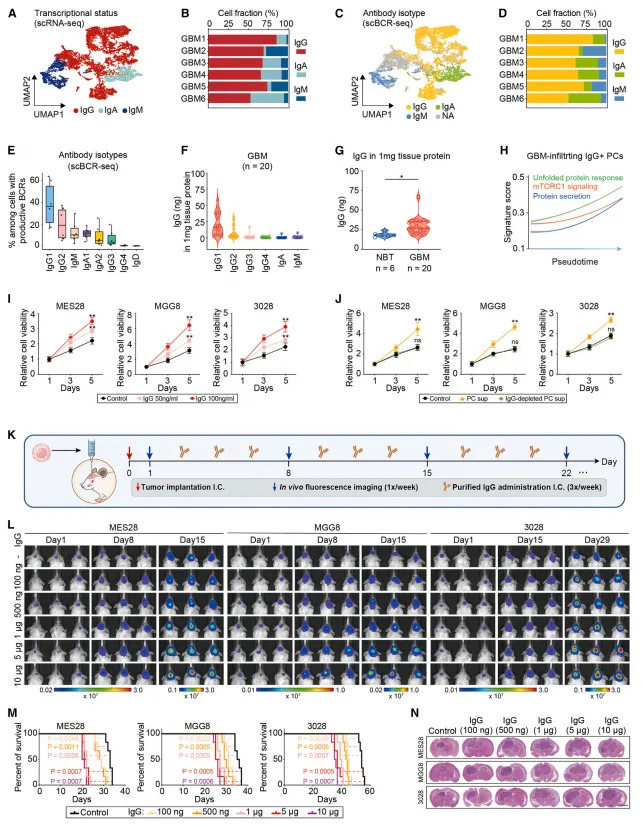
筛在各类免疫球蛋白中,仅IgG能促进胶质瘤干细胞生长,IgA和IgM无效【图4F-I】。
去除浆细胞上清中的IgG后,其促增殖效应消失。直接向颅内注射纯化IgG,足以加速肿瘤进展并恶化小鼠生存【图4J-N】。
💡锁定候选分泌因子后,必须做“添加”和“去除”双向验证,这样结论才站得住脚。
图4:浆细胞来源的IgG是驱动胶质瘤干细胞生长的关键分子,而非IgA或IgM。
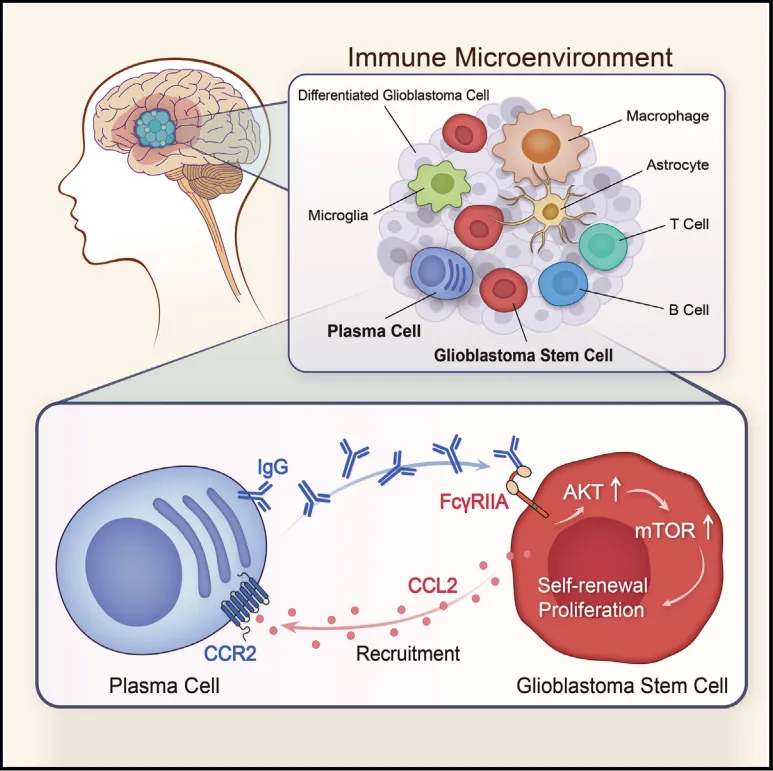
IgG结合FcγRIIA并激活AKT-mTOR通路
胶质瘤干细胞表面特异性高表达IgG受体FcγRIIA(CD32a),分化后的胶质瘤细胞则不表达【图5A】。
IgG刺激后快速诱导AKT和mTOR磷酸化,呈时间和剂量依赖性。敲低FcγRIIA后,IgG或浆细胞上清的促增殖和信号激活效应均被阻断【图5D-F】。
💡验证信号通路要遵循“激动剂+敲低/抑制剂”的逻辑,看到磷酸化变化后再反向确认,因果关系才清晰。
图5:胶质瘤干细胞特异性高表达IgG受体FcγRIIA,该受体介导IgG诱导的AKT-mTOR信号激活。
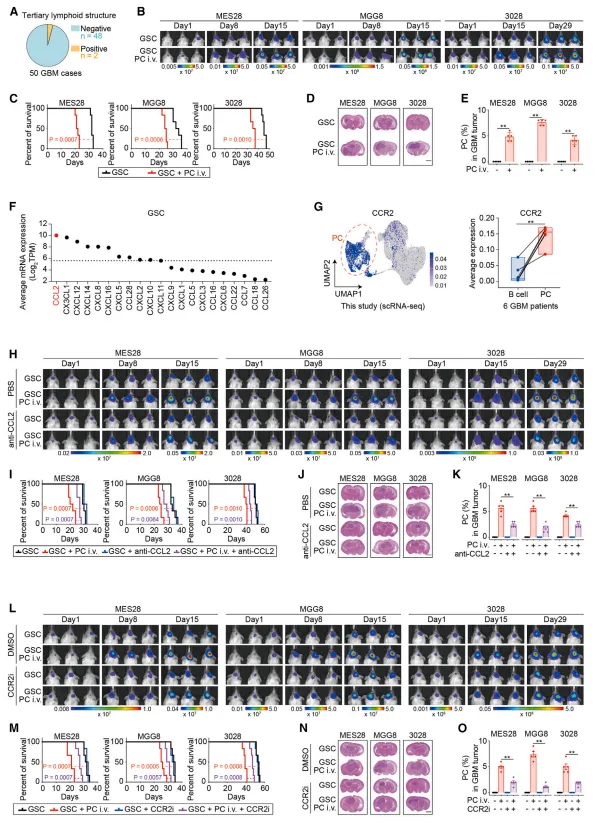
胶质瘤干细胞高表达趋化因子CCL2,而浆细胞表面高表达其受体CCR2。体外趋化实验证实,胶质瘤干细胞培养上清能有效吸引浆细胞迁移,阻断CCL2或CCR2可显著抑制这一过程【图6A-F】。💡证明A细胞促进B细胞功能后,要反过来问:B细胞怎么跑到A细胞身边的?做趋化实验和配体-受体分析就能补全这个逻辑闭环。
图6: 胶质瘤干细胞通过分泌CCL2趋化因子,将表达CCR2的浆细胞招募至肿瘤微环境。
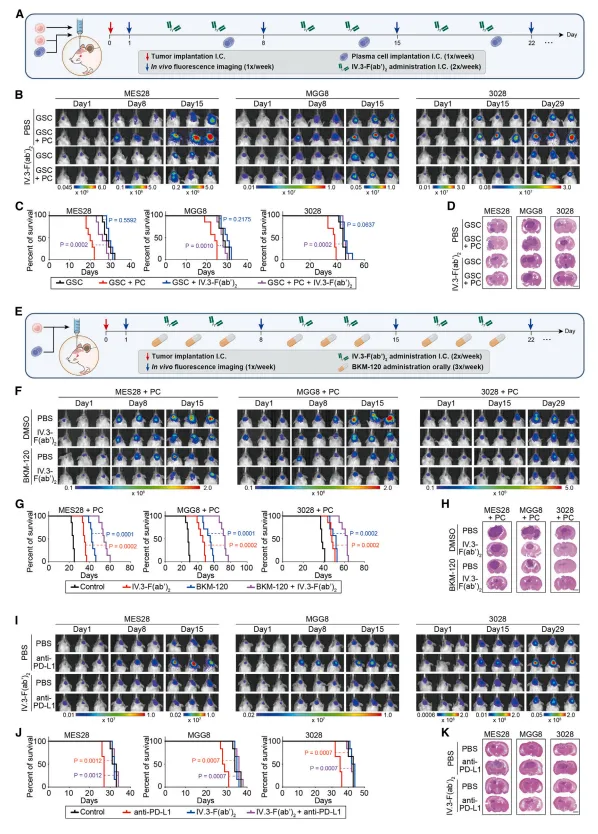
使用FcγRIIA阻断抗体(IV.3) 处理胶质瘤干细胞,不仅能抑制其增殖和成球能力,还能阻断IgG诱导的AKT-mTOR活化【图7A-E】。体内实验中,IV.3单用或联合PI3K抑制剂均能显著抑制颅内肿瘤生长并延长小鼠生存期【图7F-K】。此外,CCR2抑制剂也能减少浆细胞浸润并延缓肿瘤进展。💡机制研究最后一定要落在“能否干预”上。找到关键靶点后,用阻断抗体或抑制剂做体内治疗实验,是向转化医学迈进的关键一步。
图7: 阻断FcγRIIA或CCR2信号轴可有效抑制胶质瘤干细胞生长并延长荷瘤小鼠生存期。





 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
