南京大学新发现:靶向 PDK1/c‑Myc/SOX10 通路,逆转髓鞘损伤缓解慢性痛
- 2026-06-25 17:32:33

本研究以神经病理性疼痛(NPP)临床治疗有限、少突胶质细胞(OL)与髓鞘在 NPP 中作用不明为切入点,先构建 CCI 小鼠 NPP 模型,发现脊髓脱髓鞘与 OL 谱系细胞 PDK1 下调;进而构建 OL 谱系特异性 PDK1 条件敲除小鼠,证实 PDK1 缺失直接诱发痛觉过敏与髓鞘生成障碍;通过 RNA‑seq、分子互作等实验,揭示 PDK1 通过调控 c‑Myc 蛋白泛素化降解,解除其对 SOX10 的转录抑制,维持 OL 分化与髓鞘完整;最后通过 AAV 敲低 c‑Myc、药物 clemastine 促髓鞘再生,均能恢复髓鞘结构、改善 NPP 症状,明确 PDK1/c‑Myc/SOX10 是 NPP 发病的关键调控轴,靶向该轴可成为 NPP 治疗新策略。
神经病理性疼痛(NPP)是躯体感觉系统损伤或疾病引发的难治性慢性疼痛,全球患病率 7%–10%,现有疗法效果有限。既往研究多聚焦神经元高兴奋性与小胶质细胞、星形胶质细胞活化,中枢髓鞘与少突胶质细胞(OL)在 NPP 中的作用被严重忽视。OL 是中枢髓鞘形成细胞,维持郎飞结结构与神经冲动跳跃式传导,创伤、神经炎症等 NPP 相关诱因常导致 OL 丢失与髓鞘损伤,引发轴突传导异常与伤害性信号紊乱。PDK1 作为 PI3K/Akt 通路核心调控激酶,参与神经发生、细胞存活等中枢发育过程,但其在 OL 中的功能及与 NPP 的关联尚未被阐明,亟需揭示 OL 内在信号调控髓鞘稳态与疼痛的分子机制,为 NPP 提供新靶点。
本研究综合运用动物模型、分子生物学、组织形态学与行为学方法:构建小鼠坐骨神经慢性压迫损伤(CCI)NPP 模型;制备 NG2‑CreERT2 介导的诱导型 OL 谱系特异性 PDK1 条件敲除(cKO)小鼠;通过 PWL、PWT、旷场、平衡木、转棒等行为学评估痛觉与运动功能;采用 Western blot、qRT‑PCR、免疫组化、TrueGold 髓鞘染色、透射电镜检测髓鞘与 OL 相关指标;利用 RNA‑seq、GO/KEGG 分析筛选差异基因;通过 Co‑IP、泛素化实验、荧光素酶报告基因验证 PDK1‑c‑Myc‑SOX10 调控关系;经立体定位注射 AAV‑sh‑Myc 敲低 c‑Myc、腹腔注射 clemastine 药物干预,开展挽救实验验证通路功能。
CCI 小鼠出现明显机械性痛觉过敏与热痛觉过敏,同侧脊髓发生显著脱髓鞘,OL 谱系细胞 PDK1 特异性下调;OL 特异性 PDK1 敲除小鼠无需损伤即可重现 NPP 样痛觉过敏,且不影响运动与瘙痒相关行为,同时伴随脊髓白质减少、髓鞘蛋白(MBP、PLP1)降低、郎飞结结构破坏;机制上,PDK1 缺失不影响 c‑Myc 转录,而是通过减少 Ser62 磷酸化与泛素化导致 c‑Myc 蛋白累积,进而抑制 SOX10 转录,阻碍 OL 分化与髓鞘生成;AAV 介导脊髓 OL 特异性 c‑Myc 敲低或 clemastine 促髓鞘再生,均可恢复 SOX10 表达、修复髓鞘结构、显著缓解 PDK1 cKO 与 CCI 小鼠的神经病理性疼痛。
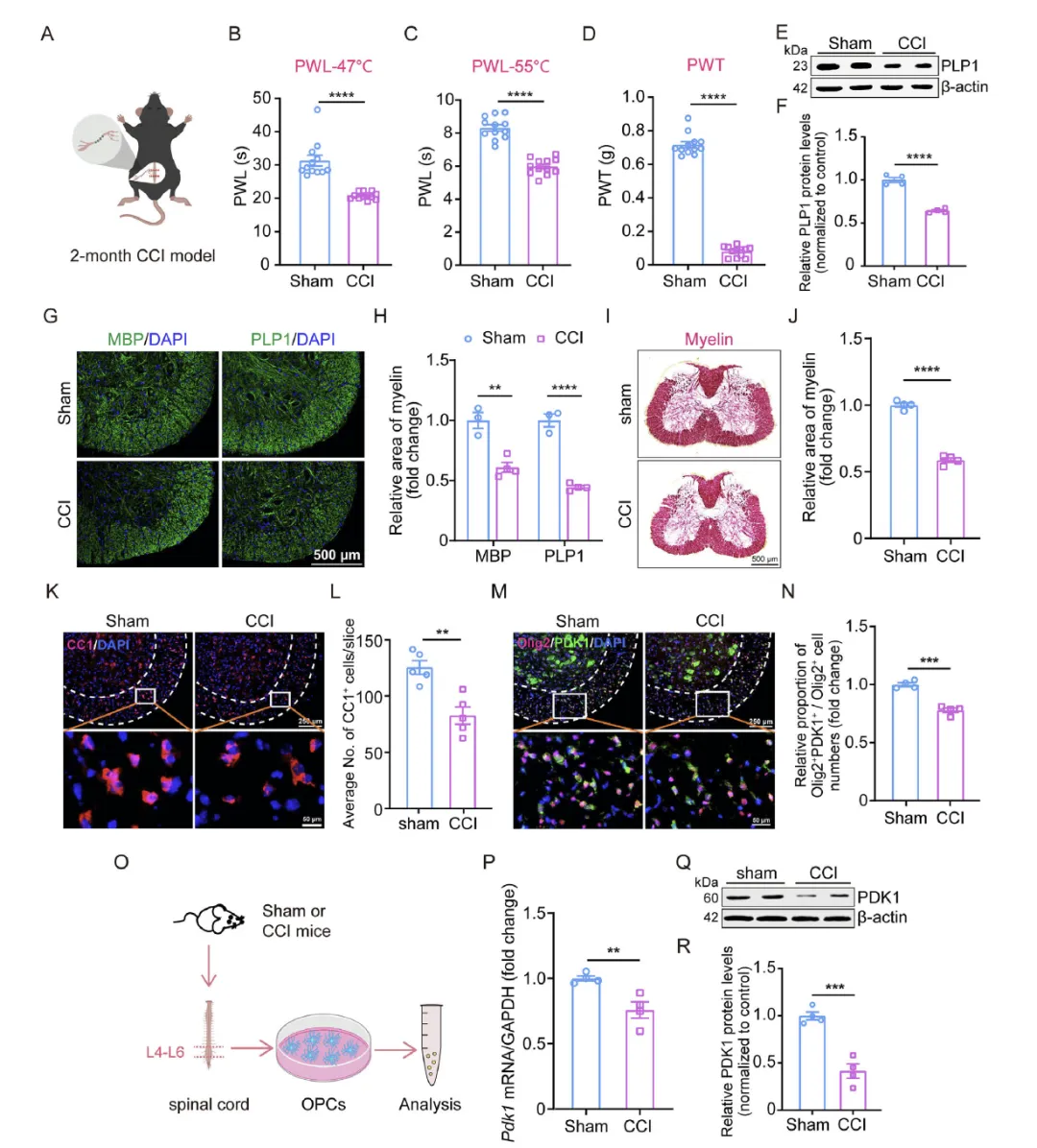
Figure 1 通过构建小鼠坐骨神经慢性压迫损伤(CCI)神经病理性疼痛模型,证实 CCI 小鼠在术后 14 天出现显著的机械性痛觉过敏与热痛觉过敏,同时同侧脊髓出现明显的髓鞘丢失、髓鞘相关蛋白(PLP1、MBP)表达降低,且少突胶质谱系细胞中 PDK1 发生特异性下调,神经元与星形胶质细胞中 PDK1 水平无变化,分离培养的少突胶质前体细胞中 PDK1 也显著降低,直接表明神经病理性疼痛伴随脊髓脱髓鞘与少突胶质细胞特异性 PDK1 下调相关。
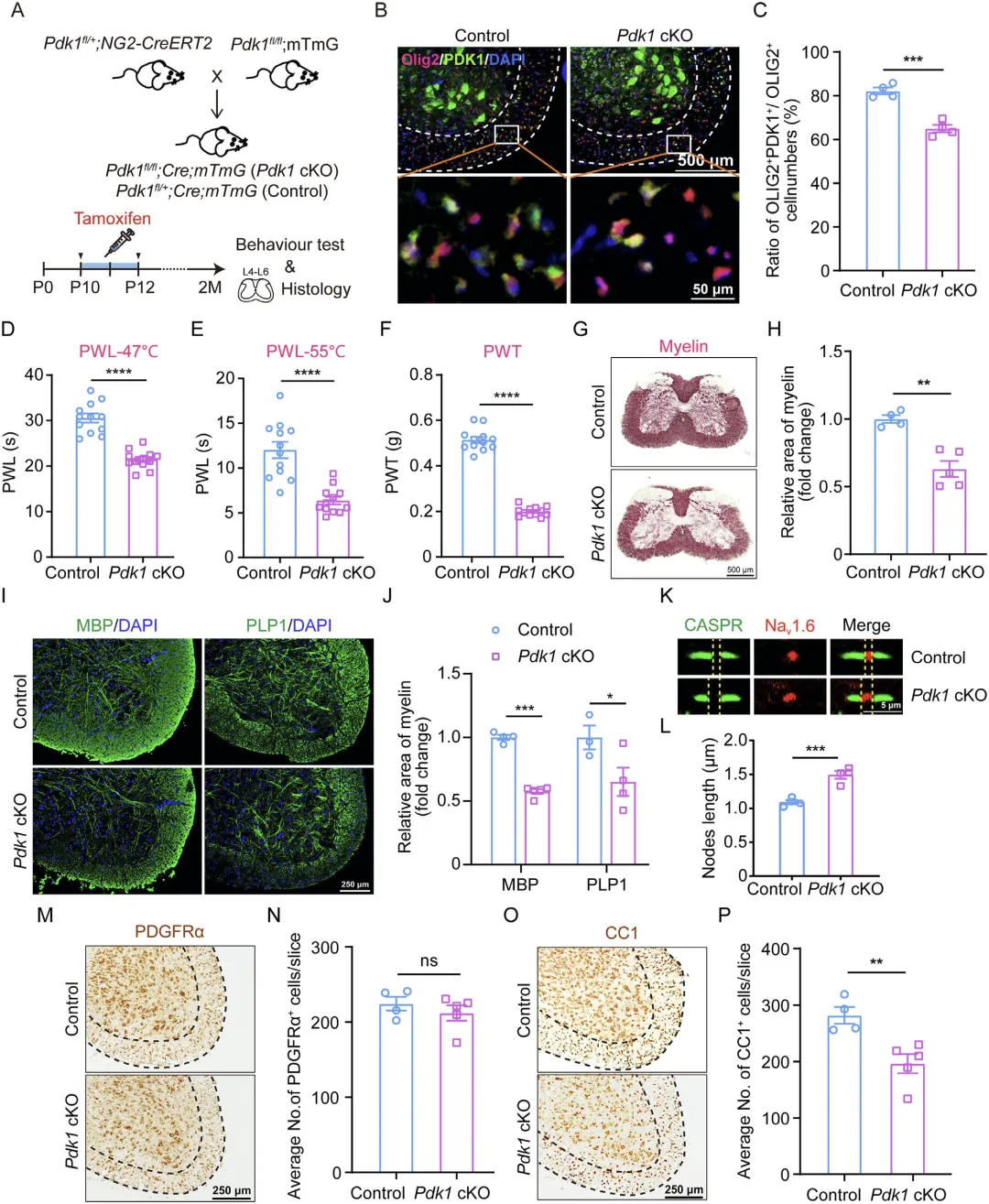
Figure 2 展示少突胶质谱系特异性 PDK1 条件敲除(Pdk1 cKO)小鼠的构建与表型,结果显示该小鼠成功实现少突胶质细胞 PDK1 特异性缺失,且无需神经损伤即可出现与 CCI 小鼠一致的机械性与热性痛觉过敏,同时运动、瘙痒相关行为无异常,脊髓出现明显白质减少、髓鞘纤维丢失、髓鞘蛋白降低、郎飞结结构破坏,成熟少突胶质细胞数量显著下降而少突胶质前体细胞数量不变,明确少突胶质细胞 PDK1 缺失直接诱发疼痛过敏与脊髓髓鞘生成障碍。
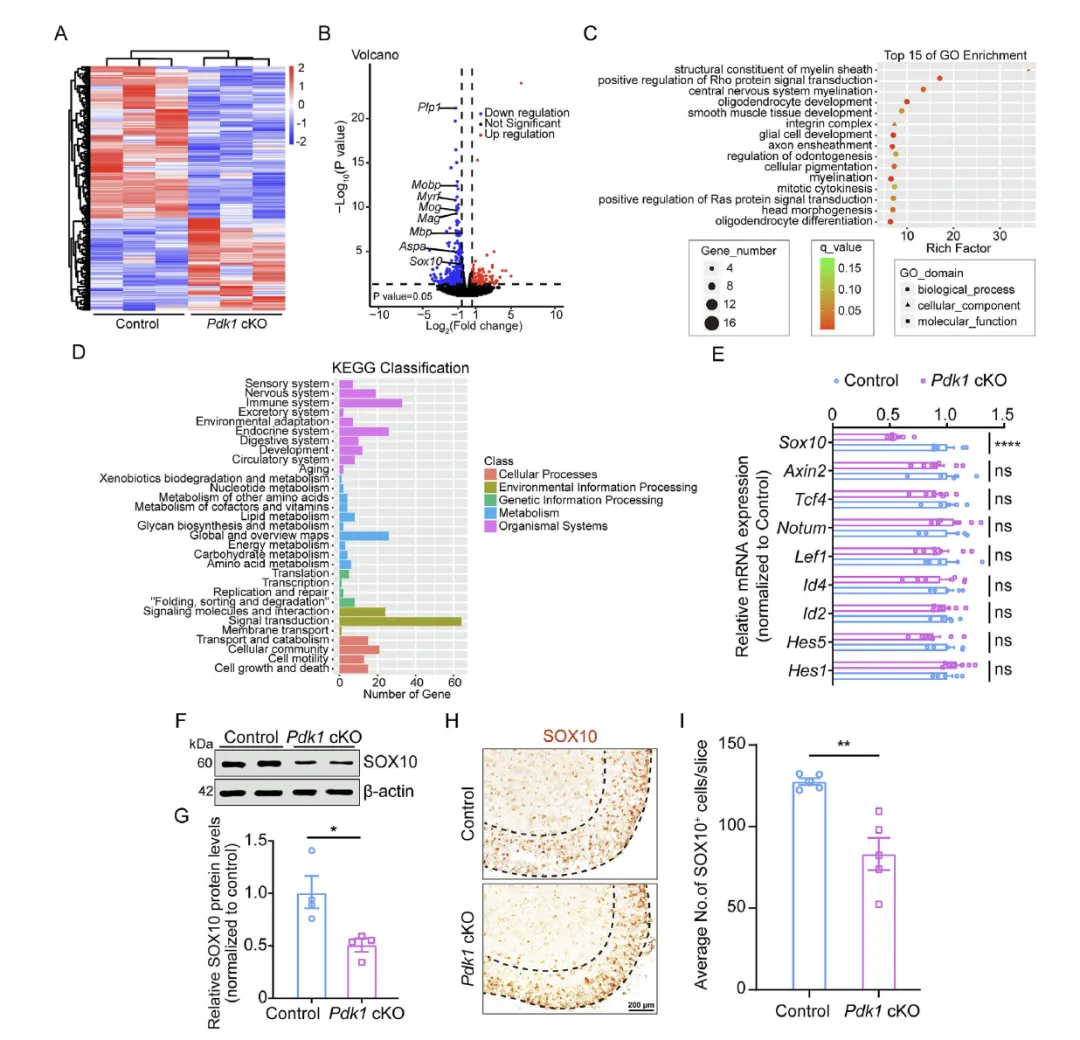
Figure 3 通过 RNA 测序、qRT‑PCR、蛋白与免疫组化检测,发现 Pdk1 cKO 小鼠脊髓中髓鞘生成与少突胶质分化关键转录因子 SOX10 显著下调,其下游髓鞘相关基因同步降低,而经典的少突胶质分化抑制因子表达无变化,GO 与 KEGG 分析显示差异基因集中于髓鞘生成与少突胶质成熟通路,证明 PDK1 缺失通过下调 SOX10 阻断少突胶质分化与髓鞘形成。
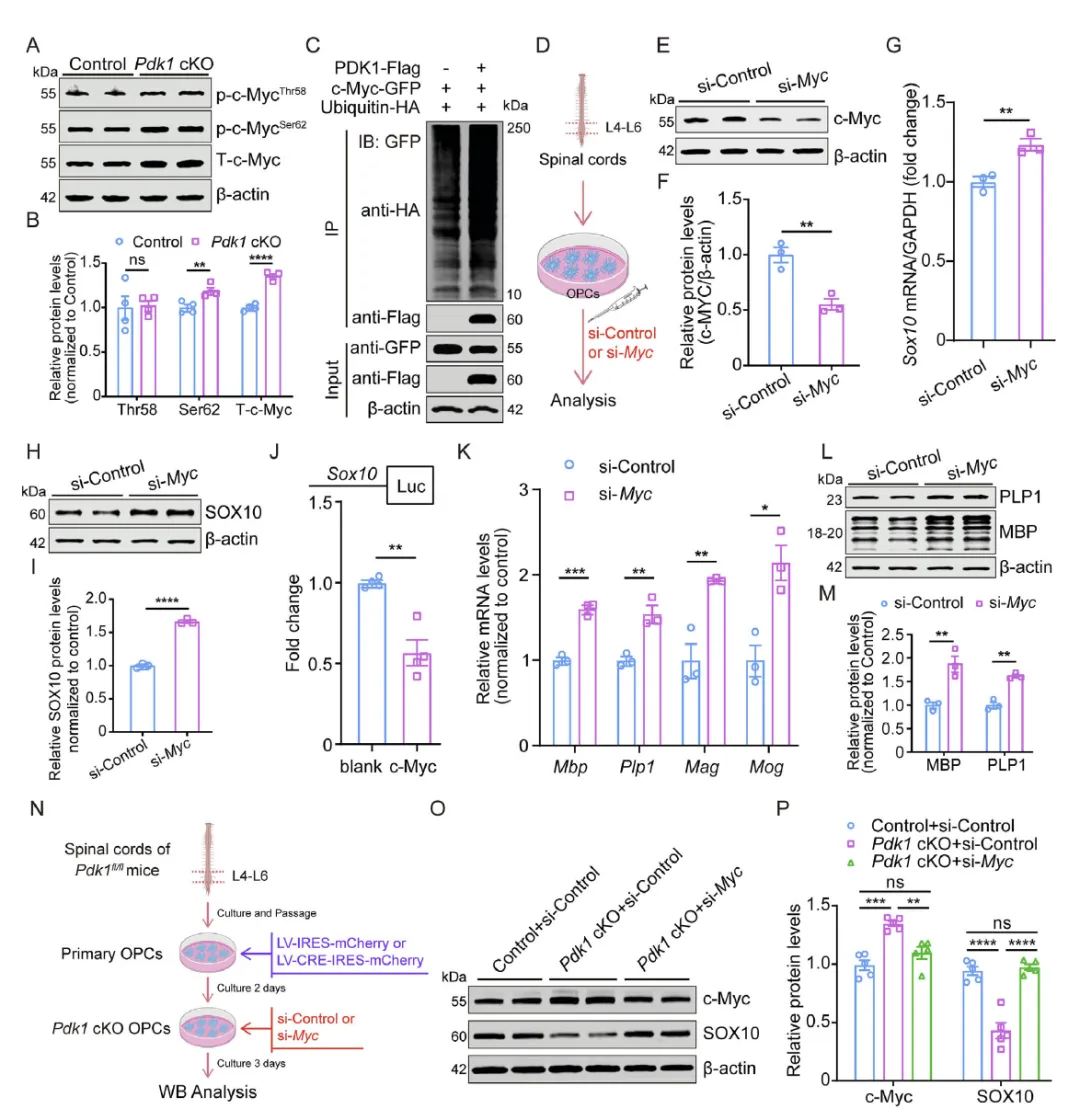
Figure 4 揭示 PDK1 调控 SOX10 与髓鞘生成的分子机制,发现 PDK1 缺失不改变 c‑Myc 的 mRNA 水平,但通过抑制 c‑Myc Ser62 位点磷酸化与泛素化降解导致 c‑Myc 蛋白累积,累积的 c‑Myc 直接抑制 Sox10 启动子活性进而下调 SOX10,敲低 c‑Myc 可恢复 SOX10 及髓鞘相关基因表达,证实 PDK1 通过泛素化调控 c‑Myc 蛋白稳定性,进而解除 c‑Myc 对 SOX10 的转录抑制以维持髓鞘生成。
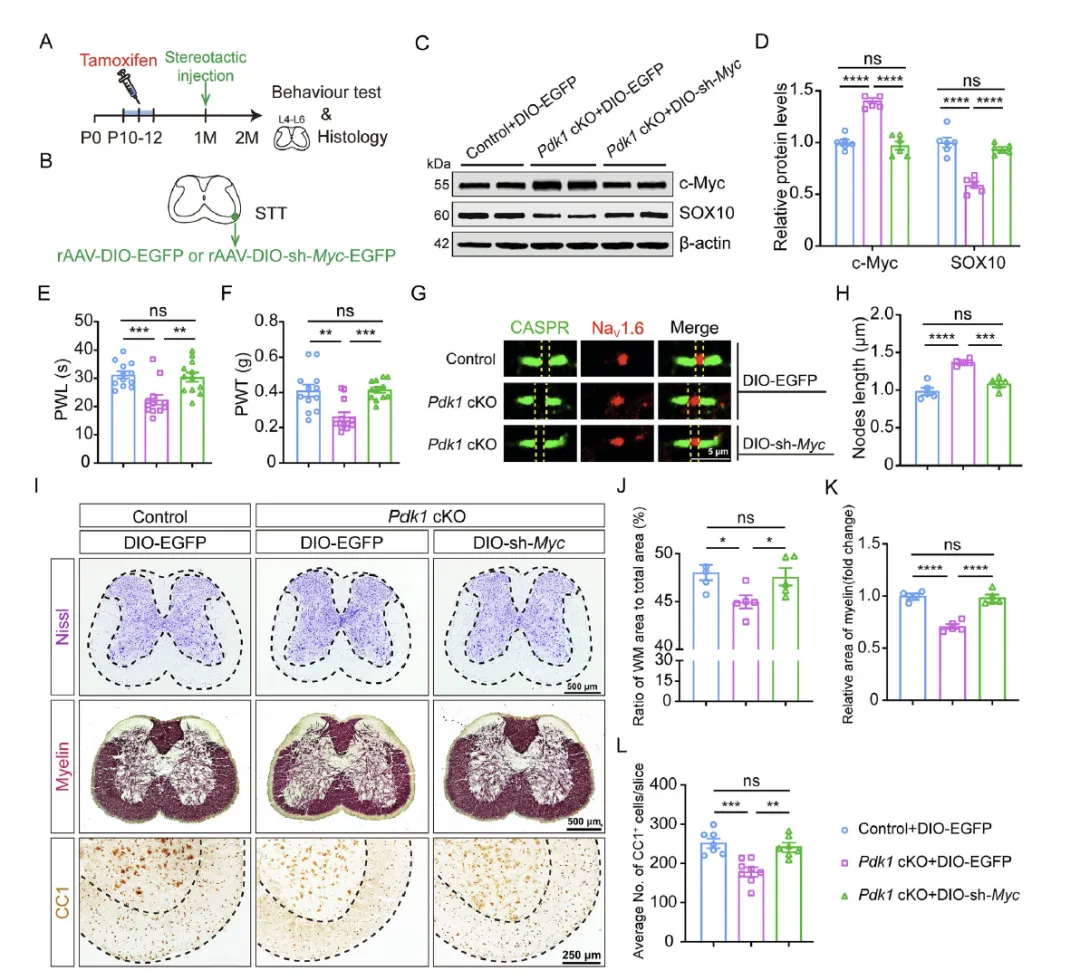
Figure 5 通过脊髓立体定位注射 AAV 介导的 Cre 依赖 sh‑Myc 敲低 Pdk1 cKO 小鼠脊髓少突胶质细胞的 c‑Myc,结果显示 c‑Myc 敲低可恢复 SOX10 表达、修复郎飞结结构、增加髓鞘纤维与成熟少突胶质细胞数量,并显著缓解 Pdk1 cKO 小鼠的机械性与热性痛觉过敏,从基因干预角度证实抑制 c‑Myc 可逆转 PDK1 缺失导致的髓鞘障碍与神经病理性疼痛。
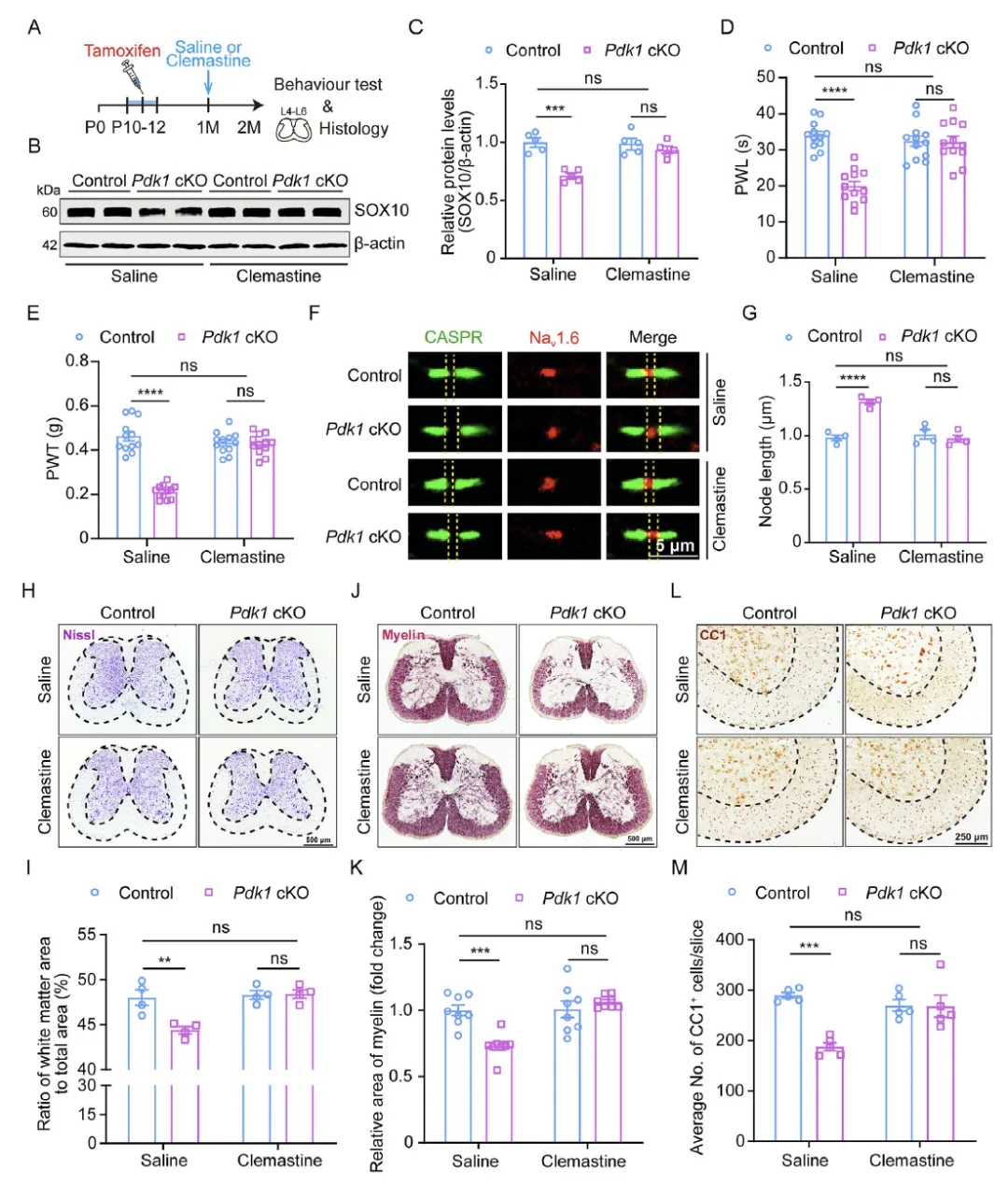
Figure 6 使用促髓鞘再生药物 clemastine 腹腔干预 Pdk1 cKO 小鼠,发现 clemastine 可恢复小鼠脊髓 SOX10 蛋白水平,修复郎飞结结构,增加白质面积、髓鞘纤维与成熟少突胶质细胞数量,并显著改善小鼠的痛觉过敏表型,从药理学角度证明促进髓鞘再生可缓解 PDK1 缺失引发的神经病理性疼痛,且该作用依赖少突胶质细胞功能完整。
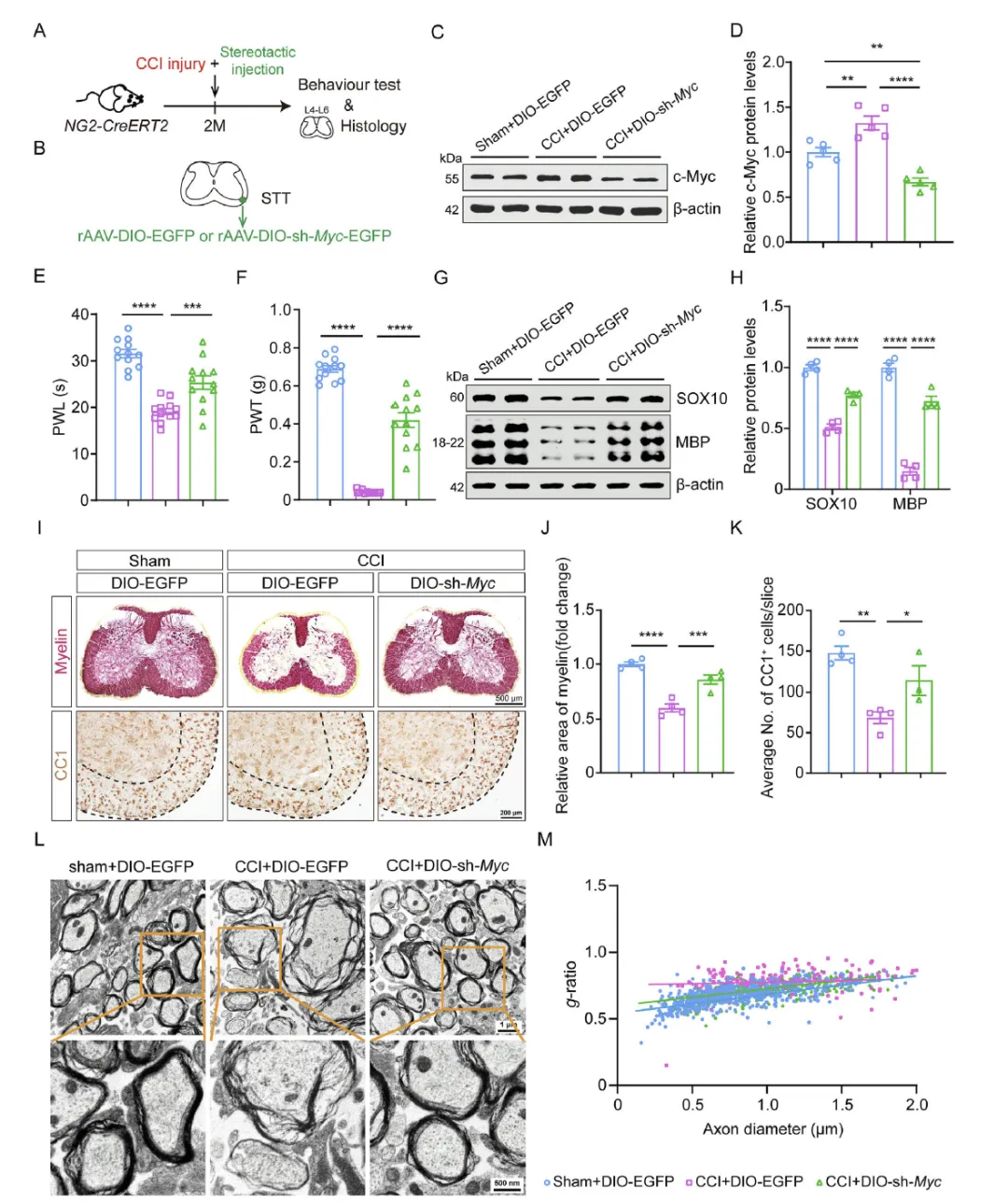
Figure 7 在 CCI 神经病理性疼痛小鼠模型中,通过 AAV 敲低脊髓少突胶质细胞的 c‑Myc,发现可降低 CCI 小鼠升高的 c‑Myc 水平,恢复 SOX10 与 MBP 表达,增加脊髓髓鞘纤维与成熟少突胶质细胞数量,超薄透射电镜显示髓鞘厚度增加、g‑比值恢复,同时显著缓解 CCI 小鼠的痛觉过敏,验证在病理疼痛模型中靶向 c‑Myc 可通过恢复髓鞘稳态缓解神经病理性疼痛。
本研究证实少突胶质细胞 PDK1 是维持中枢髓鞘稳态与抵抗神经病理性疼痛的关键分子,PDK1 通过调控 c‑Myc 蛋白降解,解除其对 SOX10 的转录抑制,保障少突胶质细胞分化与髓鞘完整;PDK1 下调引发 c‑Myc 累积、SOX10 受抑,导致髓鞘生成障碍、郎飞结破坏与脊髓感觉环路异常,最终诱发并维持神经病理性疼痛;靶向少突胶质细胞 PDK1/c‑Myc/SOX10 轴,通过基因敲低 c‑Myc 或药物促进髓鞘再生,可有效缓解神经病理性疼痛,为临床治疗提供全新机制与靶点。
局限:仅基于小鼠模型,未在人 NPP 组织验证通路;未探索 PDK1 与 ERK/NF‑κB 等疼痛通路的交叉;仅研究幼年诱导敲除,未评估成年敲除表型。展望:在人样本验证通路;拓展至糖尿病、化疗性周围神经病模型;研发靶向 c‑Myc/PDK1 的小分子镇痛药物。
国家杰青答疑实录(1对1视频)
医学省自然申请答疑,立项的关键条件是哪一些?从哪些方向可以杀出重围
临床型博士如何准备国青标书?没有预实验怎么办?专家一对一解答规划
技术精讲 & 真实案例
随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- 招聘信息 | 南京邮电大学2026年公开招聘专职辅导员(硕士、博士)公告
- 南京家长必读:大学生家教一对一辅导如何让孩子成绩飞跃?
- 南京启示录026:是阴森恐怖还是庄严肃穆
- 2026国缘四开·南京林丹杯8月开赛!比赛信息→
- 美景 南京鸡鸣寺
- 粤见南大 梦启金陵 | 南京大学2027级研究生招生宣讲咨询会【广州/深圳(含港澳专场)】报名通道开启!
- 万里云端 “英”你不凡 | 南京航空航天大学第六届飞行英语竞赛决赛顺利举行
- 唯一通讯单位!南京师范大学唐亚文/王佳丽/王宇孙瀚君,最新AFM丨物相演变造 Ru 单原子,高效碱解析氢!
- 南京市政协张宝娟主席调研江苏新联之家,走访全国政协委员、江苏省新联会会长魏青松
- 招聘信息 | 南京科技职业学院2026年公开招聘工作人员公告(第二批)
