在此,作者报道了一种酸介导的策略,用于原位1T→2H相转变的MoSe2,以构建面向碱性析氢反应的Ru位点(Ru-2H(PT)-MoSe2/CC),与直接在静态2H相MoSe2上构建Ru位点(Ru-2H-MoSe2/CC)形成对比。X射线光电子能谱证明,尽管两种催化剂具有相同的2H相结构,但相演化后的Ru-2H(PT)-MoSe2/CC在Mo和Se位点表现出显著的电子积累,使其能够与Ru3⁺前驱体产生强静电相互作用,从而决定了Ru的最终原子构型。结合理论和实验研究表明,这种强MSI优先将Ru稳定为Mo位点的单原子,同时形成较小的Ru团簇。形成鲜明对比的是,Ru-2H-MoSe2/CC中较弱的MSI导致主要形成较大的Ru团簇。所得的Ru-2H(PT)-MoSe2/CC在10mAcm⁻2时具有26mV的低过电位,并在100mAcm⁻2下具有500h的长期耐久性。原位拉曼光谱和理论计算进一步证实,独特的Ru构型优化了界面水行为并加速了水解离动力学。碱性电催化析氢反应代表了一种大规模生产氢气(H2)的环保方法。铂(Pt)因其适中的氢吸附能(ΔG*H)而被公认为基准催化剂,但其高成本仍然是一个限制因素。钌(Ru)具有高活性和相对较低的价格,已成为一种有前景的替代物,人们已开发出各种策略来进一步提高其本征活性并降低金属负载量。
其中,构建负载型多相催化剂已被证明在提高本征活性和最小化Ru用量方面有效。值得注意的是,除了作为分散Ru物种的载体外,载体还可以通过强金属-载体相互作用(MSI)调节Ru的性质。
因此,鉴于载体材料的关键作用,人们已探索了多种方法,包括组成调控、形貌控制、尺寸调控和相工程,以阐明Ru位点与载体之间的结构-性能关系。特别是,相工程提供了一条调控活性位点原子排列、配位环境和电子结构的基本途径,从而直接影响金属物种的原子构型和最终的MSI。
作为电催化剂载体的重要成员,过渡金属二硫族化合物(TMDs)表现出高度依赖于其晶体相的电子结构。常规的2H相具有优异的热力学稳定性,但其惰性的基面导致与负载金属的电子相互作用较弱。
相比之下,1T相在基面上含有丰富的缺陷位点,能够实现与金属物种更强的电子耦合,但其热力学不稳定,在操作条件下容易发生相变。这种相依赖的相互作用已在近期研究中得到阐明。
例如,Pt的单原子分散已在1T相TMDs上实现,而Pt纳米颗粒的外延生长通常发生在2H相上,前者表现出更优异的HER活性。尽管取得了这些进展,目前对MSI的理解主要基于静态相结构,而通过原位相变对MSI进行动态操控,特别是其在指导金属位点原子构型方面的作用,很少被探索。
在此,作者报道了一种新颖的酸介导策略,以触发MoSe2中的原位1T→2H相转变,从而构建独特构型的Ru基催化剂。所得催化剂(Ru-2H(PT)-MoSe2/CC)与通过常规Ru沉积在静态2H-MoSe2载体上制备的催化剂(Ru-2H-MoSe2/CC)进行了比较。
虽然X射线衍射(XRD)和拉曼光谱证实两种催化剂具有相同的最终2H相,但X射线光电子能谱(XPS)揭示,相演化后的Ru-2H(PT)-MoSe2/CC在Mo和Se位点表现出显著的电子积累。这种电子再分配促进了与Ru3⁺前驱体的强静电相互作用,并最终决定了Ru物种的原子级构型。
结合X射线吸收光谱(XAS)和从头算分子动力学(AIMD)模拟表明,在Ru-2H(PT)-MoSe2/CC中,Ru主要以单原子形式取代Mo位点,同时形成小的Ru团簇,而在静态对应物中则构建了较大的Ru团簇。借鉴先前文献中建立的方法,作者对结构和性能差异的多种影响因素进行了系统解耦。基于Brunauer-Emmett-Teller(BET)比表面积分析和电子顺磁共振(EPR),排除了形貌和空位的潜在决定性作用。
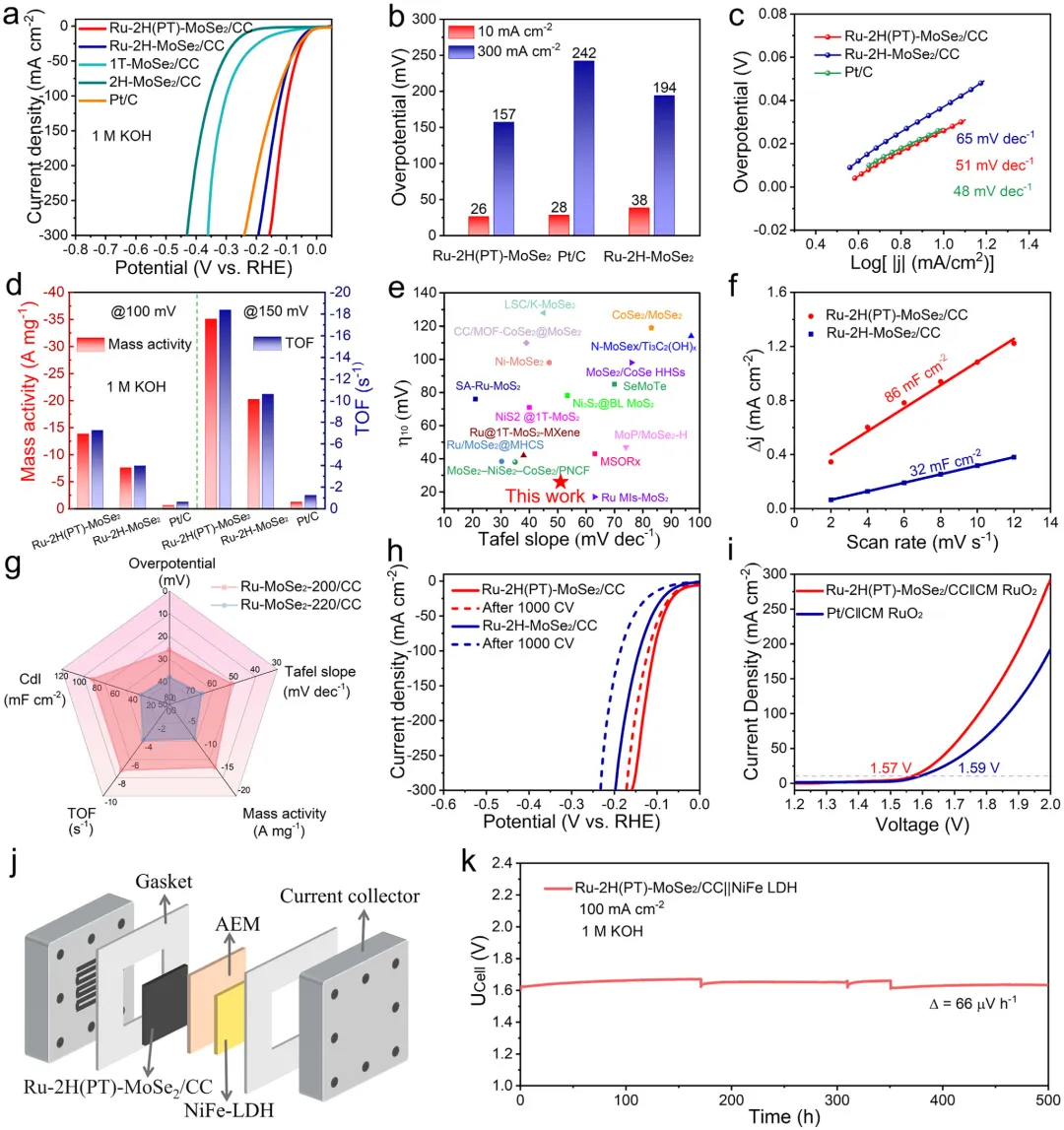
因此,可以得出结论,原位相演化过程控制了Ru-2H(PT)-MoSe2/CC中Ru的构型,这归因于Ru与原始1T-MoSe2之间有利的能级匹配,从而增强了MSI。因此,Ru-2H(PT)-MoSe2/CC展现出优异的HER性能,在10mAcm⁻2时过电位为26mV,在100mAcm⁻2下具有超过500h的长期耐久性。
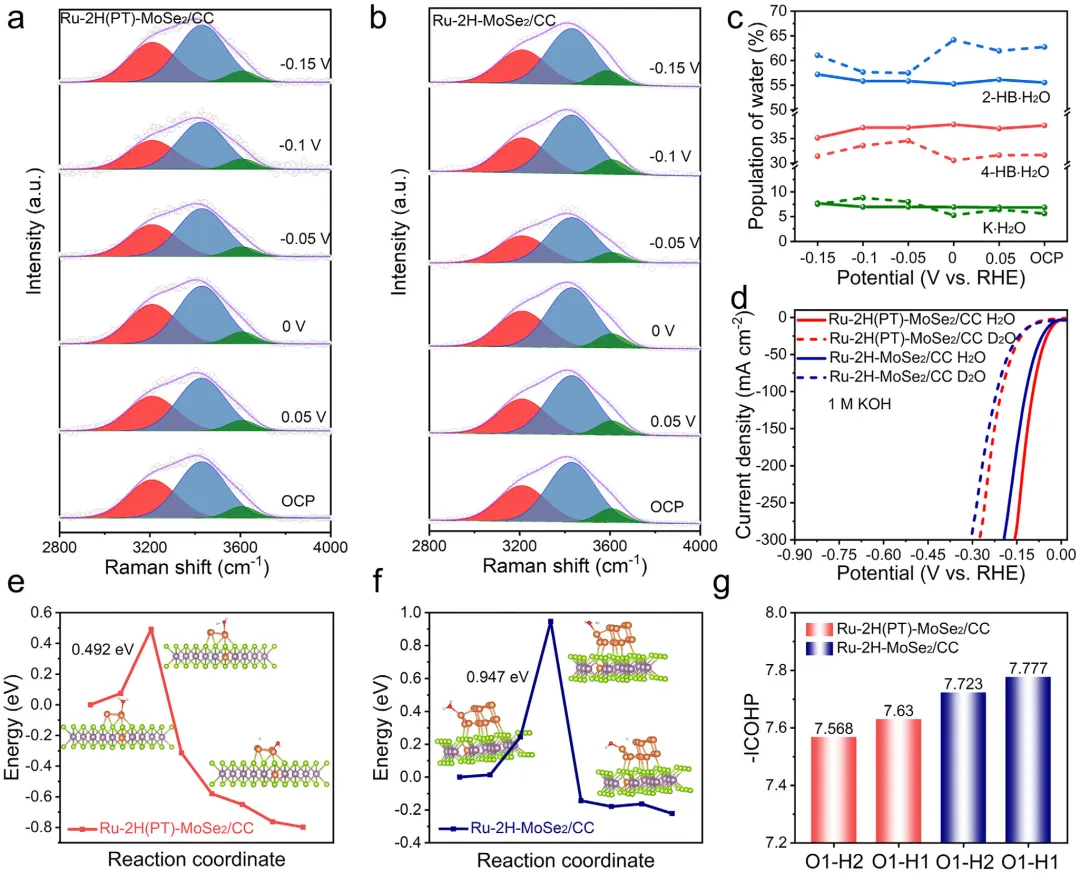
通过原位拉曼光谱、动力学同位素效应(KIE)分析和密度泛函理论(DFT)计算,作者进一步验证了水解离是速率决定步骤,并且Ru-2H(PT)-MoSe2/CC表现出增强的水分解能力,从而加速了整体反应动力学。
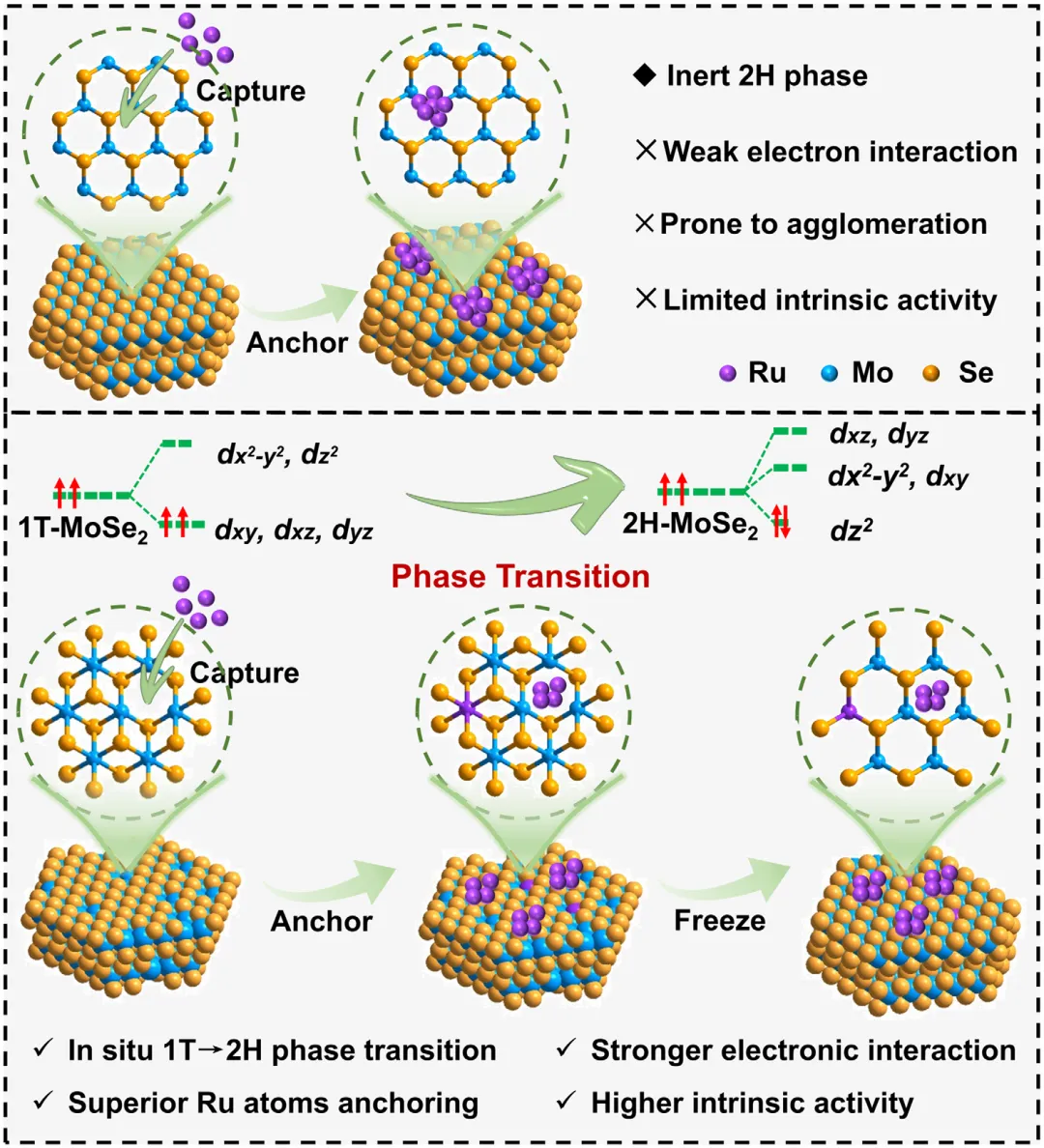
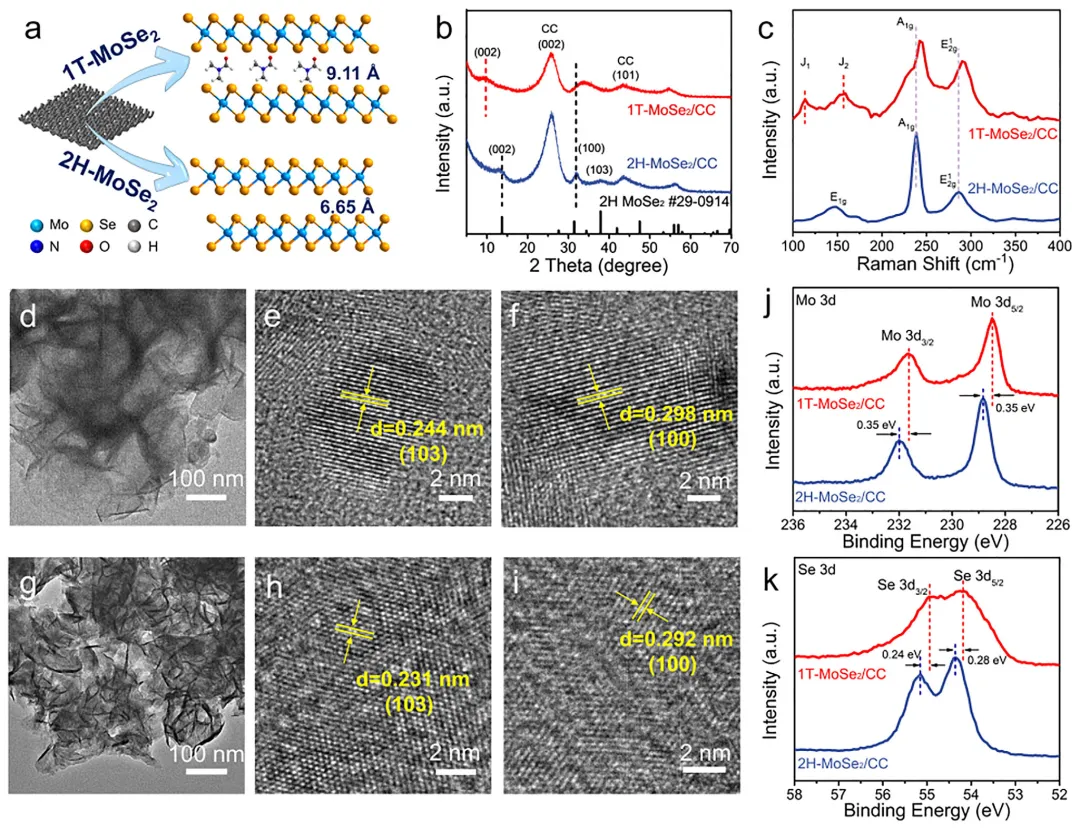
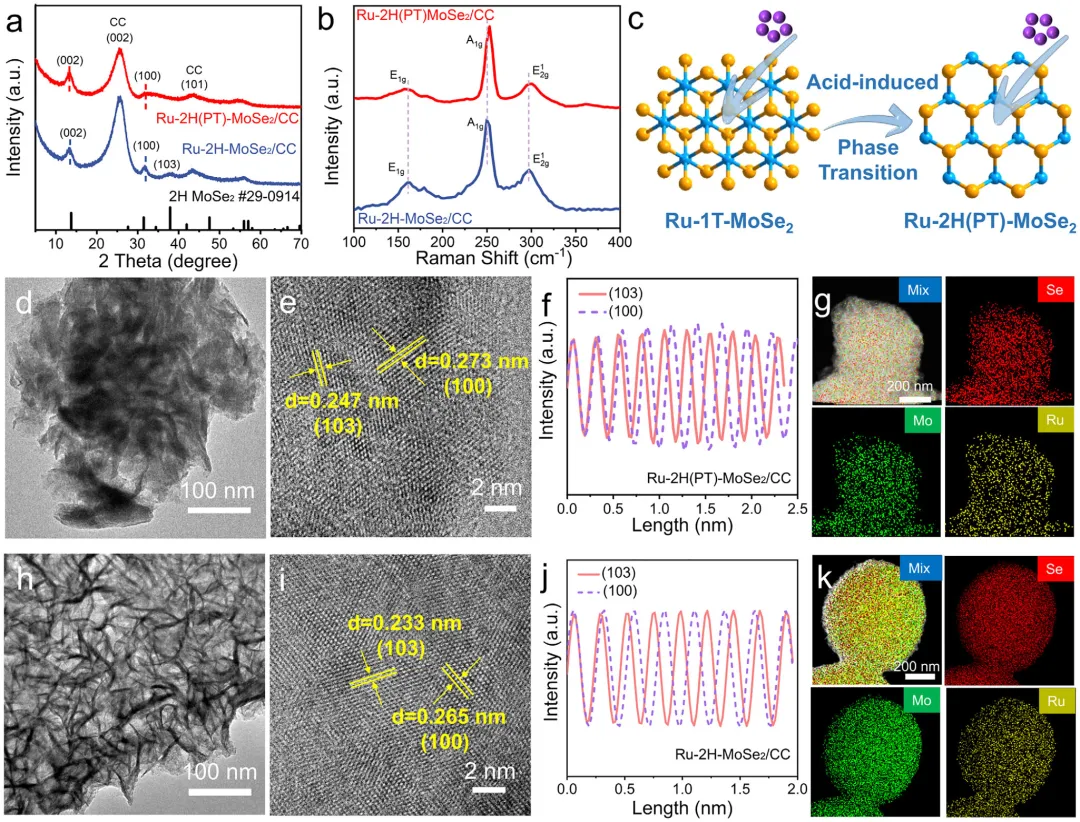
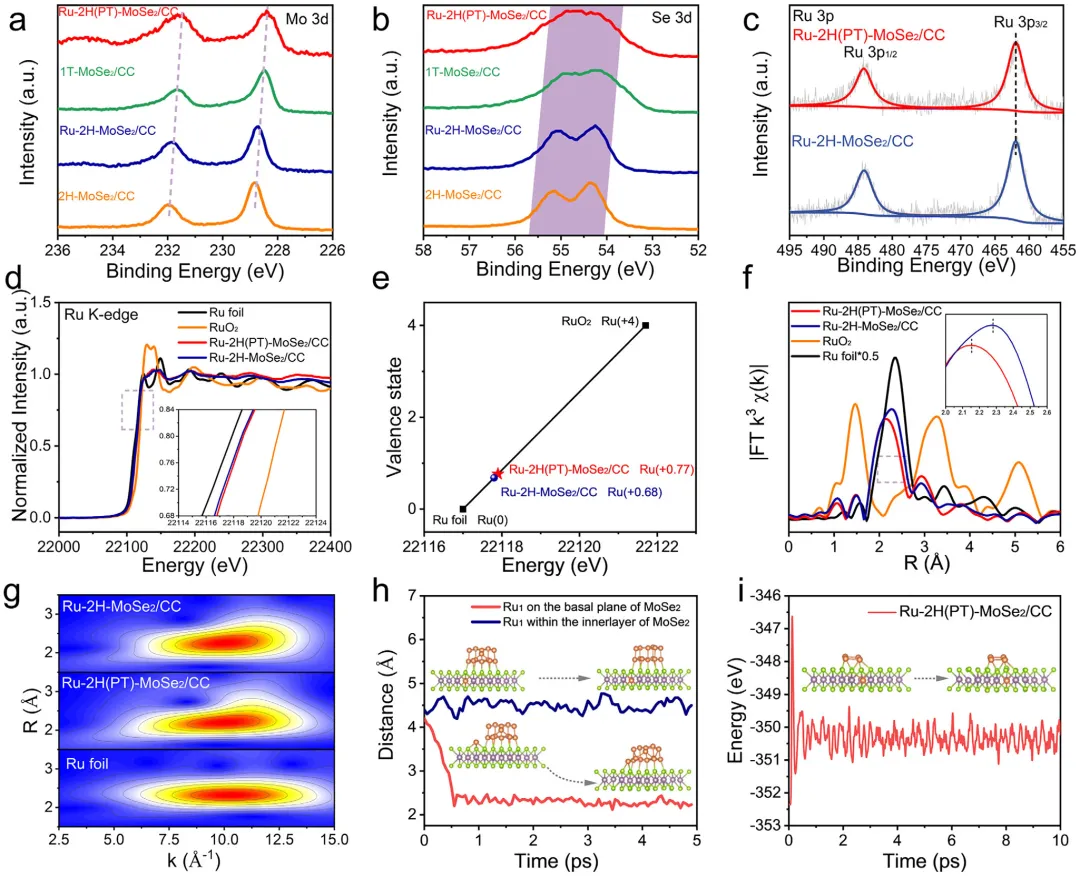
图1:原位相转变效应对调控结构和电子态的示意图。该图展示了原位相转变对调控结构和电子态的影响机制,对比了静态2H相上Ru的弱电子相互作用、易团聚和有限本征活性,与通过原位1T→2H相转变实现的强电子相互作用、优异的Ru原子锚定和更高本征活性。图2: MoSe2/CC的结构表征。(a)1T和2H-MoSe2/CC合成示意图;(b)1T和2H-MoSe2/CC的XRD图谱;(c)1T和2H-MoSe2/CC的拉曼光谱;(d-f)1T-MoSe2/CC的TEM和HRTEM图像;(g-i)2H-MoSe2/CC的TEM和HRTEM图像;(j,k)1T和2H-MoSe2/CC的Mo3d和Se3dXPS光谱。图3: Ru-2H(PT)-MoSe2/CC和Ru-2H-MoSe2/CC的结构表征。(a)XRD图谱;(b)拉曼光谱;(c)Ru-2H(PT)-MoSe2/CC合成过程中相转变示意图;(d-g)Ru-2H(PT)-MoSe₂/CC的TEM、HRTEM、积分像素强度和EDS元素分布图像;(h-k)Ru-2H-MoSe₂/CC的TEM、HRTEM、积分像素强度和EDS元素分布图像。图4:催化剂的XPS和XAFS光谱。(a)Mo3d的XPS光谱;(b)Se3d的XPS光谱;(c)Ru3p的XPS光谱;(d)Ru箔、Ru-2H(PT)-MoSe2/CC、Ru-2H-MoSe2/CC和RuO2的RuK边XANES光谱(插图:Ru箔、Ru-2H(PT)-MoSe2/CC、Ru-2H-MoSe2/CC和RuO2的不同吸收能(E0)的RuK边);(e)Ru箔、Ru-2H(PT)-MoSe2/CC、Ru-2H-MoSe2/CC和RuO2的RuK边吸收能(E0)与氧化态的关系;(f)RuK边FT-EXAFS光谱;(g)RuK边Ru箔、Ru-2H(PT)-MoSe2/CC和Ru-2H-MoSe2/CC的WT图;(h)AIMD模拟期间位于基面(吸附Ru单原子)和层间(取代Ru单原子)的Ru单原子(Ru1)与Ru团簇之间Ru-Ru距离的评估(插图:初始态和最终态的侧视图);(i)Ru-2H(PT)-MoSe2/CC在10psAIMD模拟期间的能量波动(插图:初始态和最终态的侧视图)。图5:1.0MKOH中催化剂的电化学分析。(a)扫描速率为5mVs⁻1的线性扫描伏安法(LSV)曲线;(b)10mAcm⁻2和300mAcm⁻2时的过电位对比;(c)Tafel图;(d)Ru-2H(PT)-MoSe2/CC、Ru-2H-MoSe2/CC和Pt/C在100mV和150mV时的质量活性和TOF对比;(e)与近期报道的HER电催化剂的对比;(f)扫描速率为2至12mVs⁻1的CV的Cdl测试;(g)雷达图对比;(h)初始状态和1000次循环后的HER极化曲线;(i)扫描速率为5mVs⁻1的整体水分解装置的LSV曲线;(j)AEMWE示意图;(k)使用Ru-2H(PT)-MoSe2/CC和NiFeLDH在100mAcm⁻²下的AEMWE稳定性测试(观察到的电位波动源于电解液更新和需要系统重启的意外断电)。图6: 该图展示了原位拉曼光谱、动力学同位素效应实验和DFT计算结果,揭示了Ru-2H(PT)-MoSe2/CC和Ru-2H-MoSe2/CC在界面水结构和水分解动力学方面的差异。(a)Ru-2H(PT)-MoSe2/CC和(b)Ru-2H-MoSe2/CC的原位拉曼光谱,三种O─H伸缩模式(νO─H)分别对应4配位氢键水(4-HB-H2O)、2配位氢键水(2-HB-H2O)和水合阳离子水(M⁺-H2O),以红色、蓝色和绿色显示;(c)原位拉曼光谱的电位依赖性界面水比例,实线:Ru-2H(PT)-MoSe2/CC,虚线:Ru-2H-MoSe2/CC;(d)Ru-2H(PT)-MoSe2/CC和Ru-2H-MoSe2/CC在KOH/H2O和KOD/D2O中的LSV曲线;(e,f)Ru-2H(PT)-MoSe2/CC和Ru-2H-MoSe2/CC的H2O解离动力学能垒,插图:Ru-2H(PT)-MoSe2/CC和Ru-2H-MoSe2/CC的初始态、过渡态和最终态;(g)吸附在Ru-2H(PT)-MoSe2/CC和Ru-2H-MoSe2/CC上的H2O的O─H键强度的ICOHP曲线。本研究通过酸介导的原位1T→2H相转变策略,成功调控了MoSe2载体与Ru物种之间的强金属-载体相互作用,实现了Ru单原子与小型Ru团簇的协同构型,显著优化了界面水行为和水分解动力学。该催化剂在碱性析氢反应中展现出26mV的极低过电位和500h的超长稳定性,并可在阴离子交换膜电解槽中稳定运行,为设计高效、低成本的非铂基碱性析氢电催化剂提供了新的相工程策略和理论指导,在绿氢制备和可再生能源存储领域具有广阔的应用前景。Dynamic Phase-Evolution-Triggered Strong Metal-Support Interaction Configures Atomic Ru Sites for Superior Alkaline Hydrogen Evolution,Advanced Functional Materials,2026,https://doi.org/10.1002/adfm.76311