『镁电』南京师范大学 & 悉尼科大团队 Angew. Chem. Int. Ed.: 离子液体阳离子氯锚定添加剂,加速脱溶动力学构筑无活化长效镁金属电池
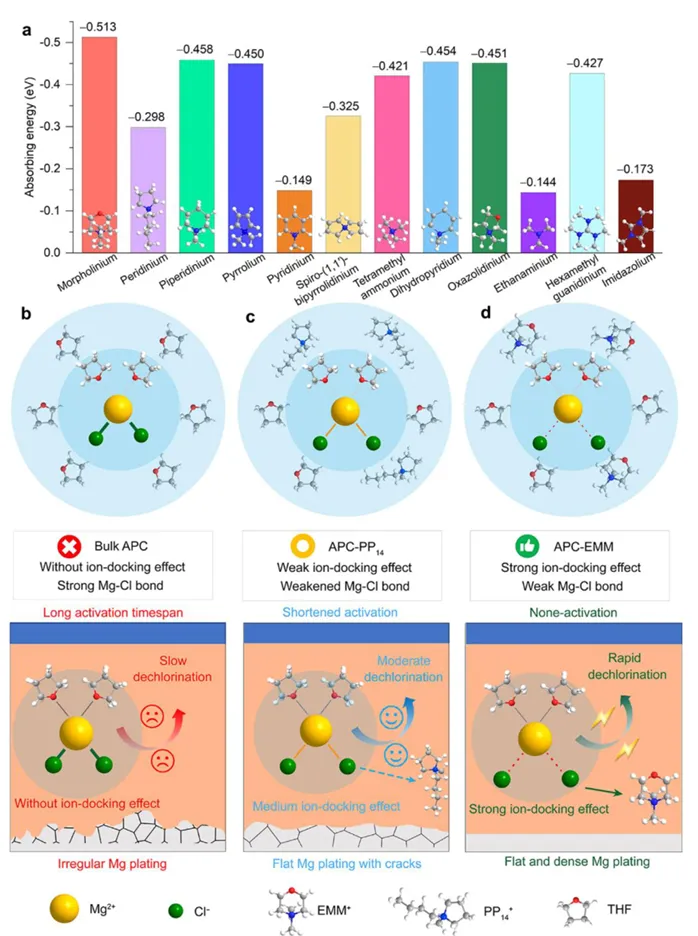
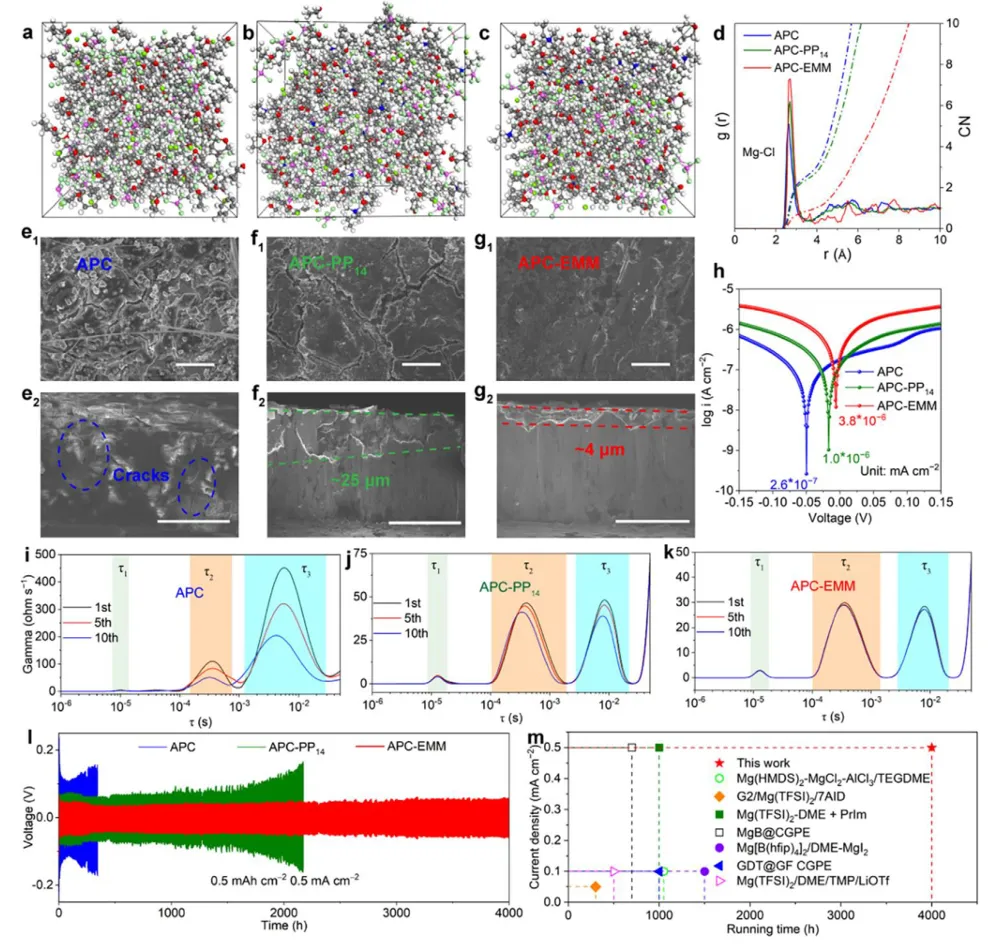
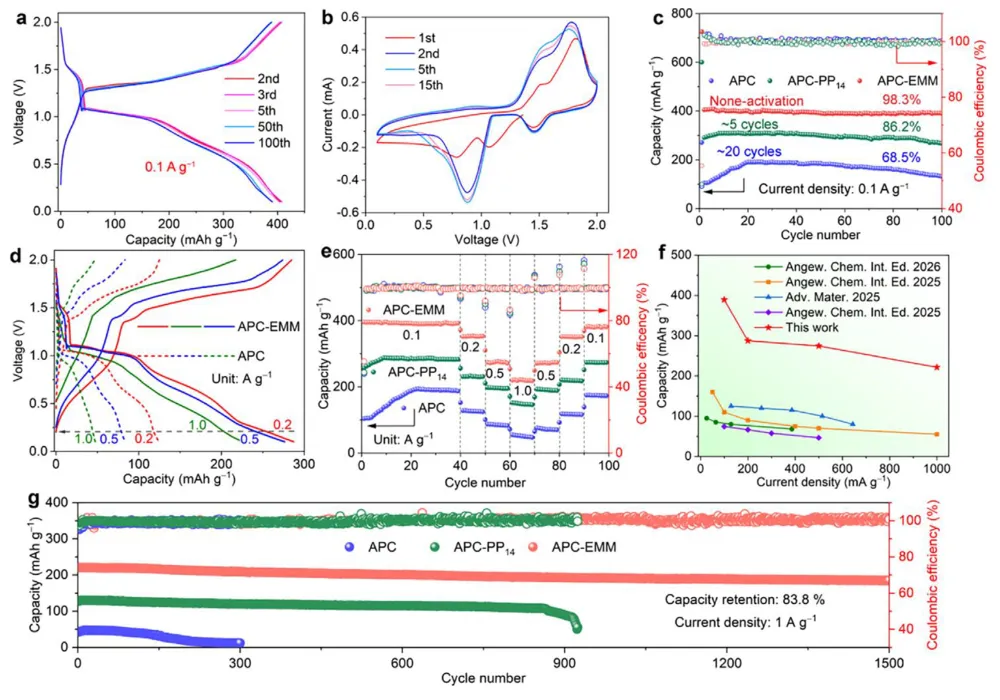
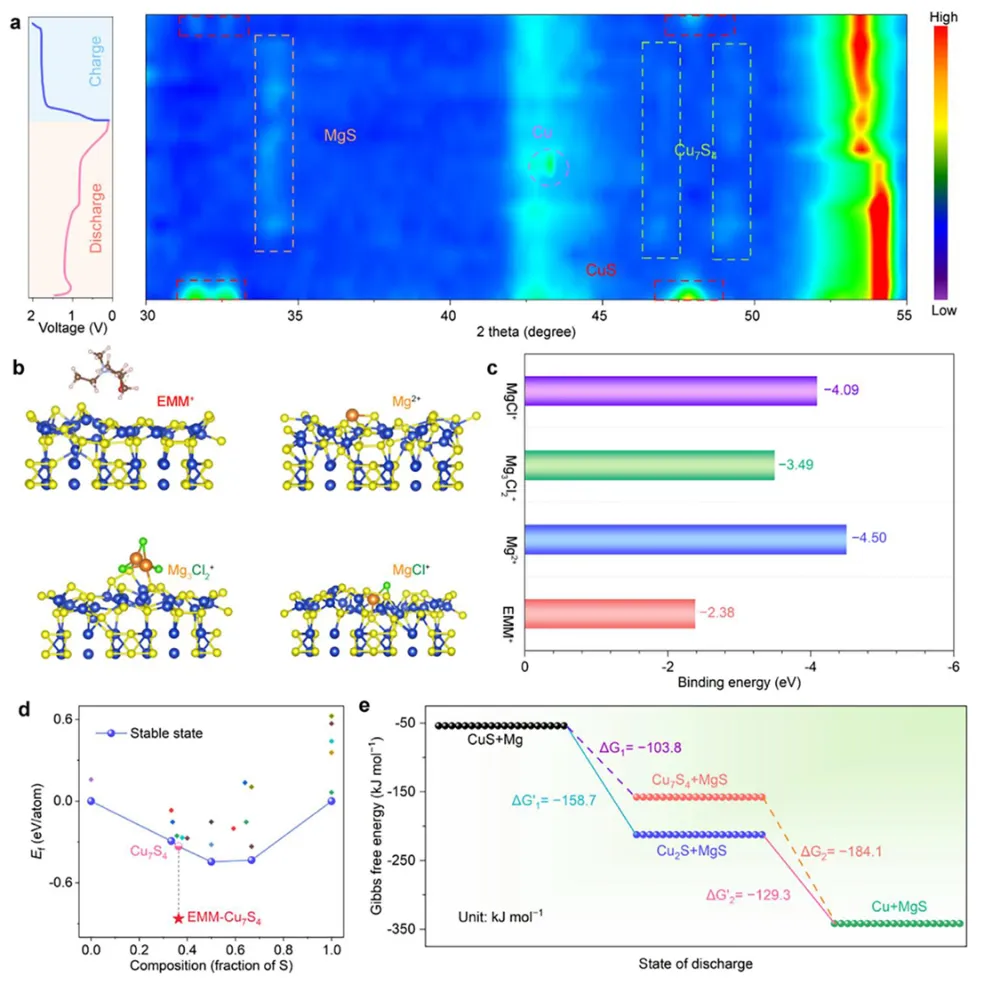
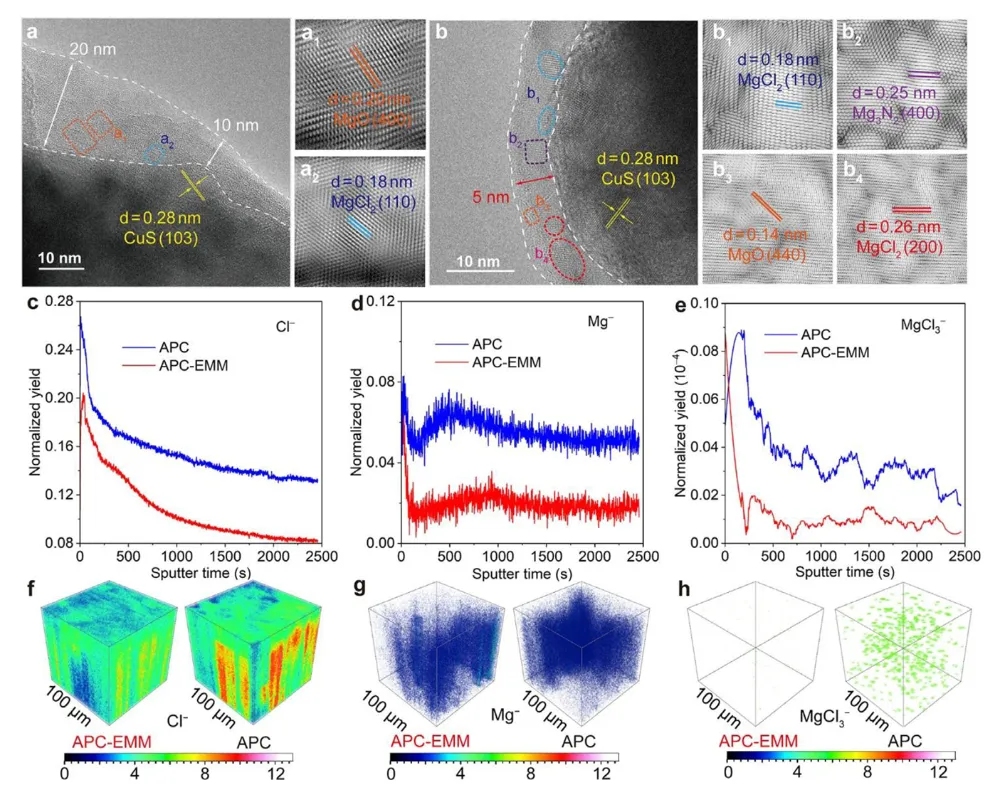
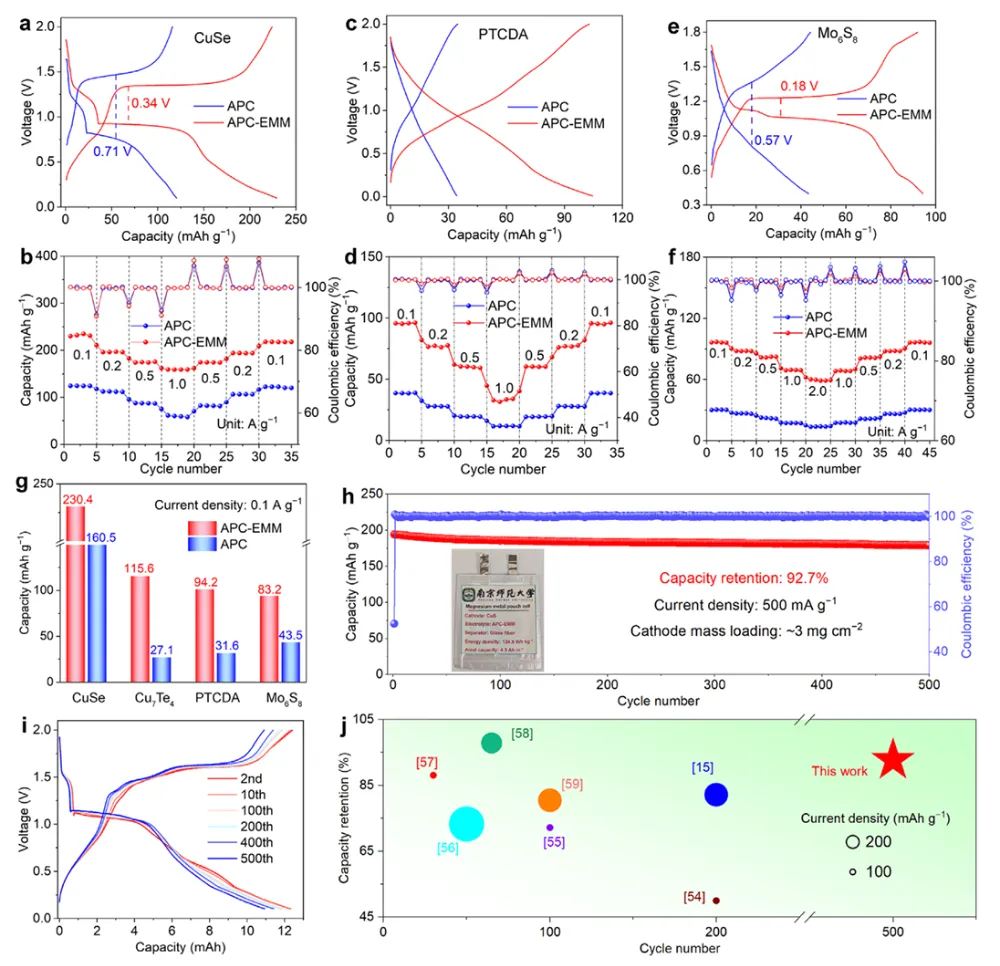
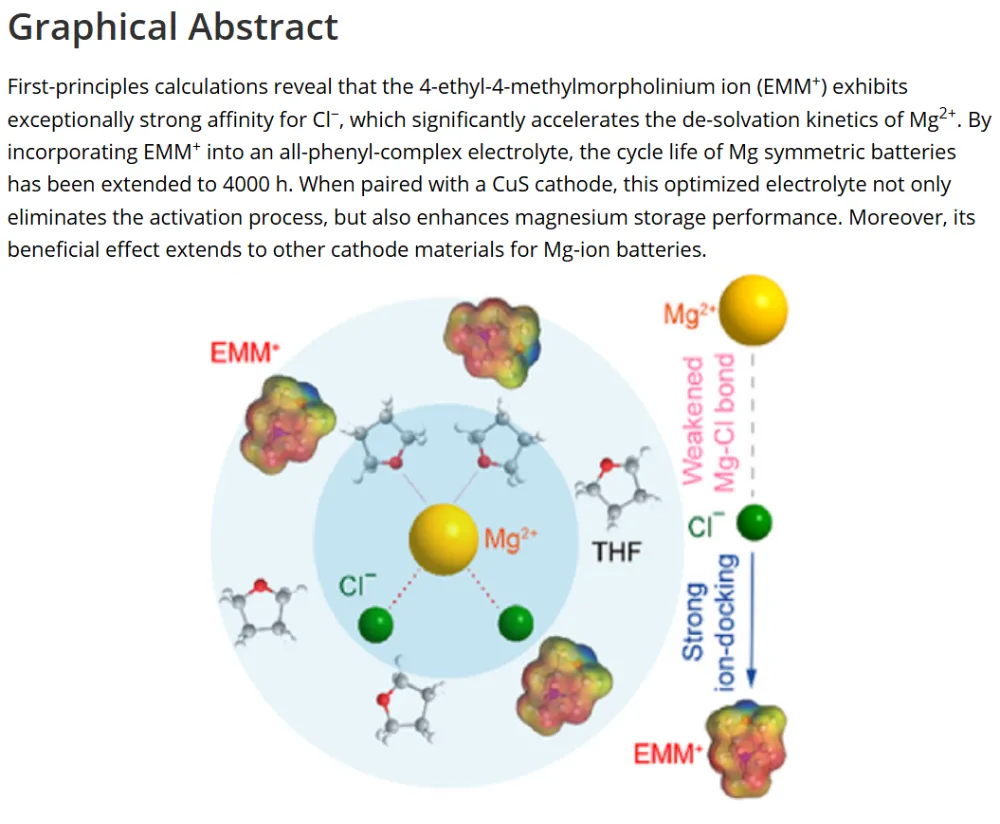
镁金属负极拥有高体积容量(3833 mAh L⁻¹)、储量丰富、本征无枝晶安全优势,是后锂储能极具潜力的技术路线。但镁离子电池产业化受多重核心难题制约:二价 Mg²⁺携带强库仑作用力,在全苯基复合(APC)电解液中极易形成稳定 Mg-Cl 配位溶剂鞘,脱溶活化势垒极高,电池需要数十圈活化周期才能达到额定容量,倍率性能严重劣化;传统 APC 电解液游离氯离子含量高,持续腐蚀镁金属负极,循环过程界面副反应不断累积。现有改性手段大多聚焦电极形貌、元素掺杂,极少从电解液溶剂化动力学角度解决脱溶限速短板;普通离子液体添加剂仅适配嵌层型正极,对转化型铜基硫化 / 硒正极提升效果微弱,同时无法同步兼顾镁负极可逆沉积、正极界面稳定两大需求。缺少可通用、低成本、能够彻底消除活化阶段的电解液调控方案,高可逆镁金属储能体系发展存在关键理论与工程瓶颈。本工作通过 DFT 理论筛选,设计 4 - 乙基 - 4 - 甲基吗啉阳离子(EMM⁺)作为氯锚定离子液体添加剂,构建 APC-EMM 复合电解液。EMM⁺与氯离子强结合(吸附能 - 0.513 eV),削弱 Mg-Cl 配位键,打散致密溶剂鞘,大幅降低 Mg²⁺脱溶能垒,彻底消除电池活化周期。EMM⁺同时调控正负极界面:负极诱导均匀平整镁沉积、抑制腐蚀开裂;正极原位生成以MgCl₂为主的薄型无机 CEI 膜,稳定 Cu₇S₄可逆中间相。选用CuS纳米片正极验证体系,0.1 A g⁻¹ 容量可达 405.1 mAh g⁻¹,1 A g⁻¹ 下循环 1500 圈容量保有 83.8%;该电解液对CuSe、Cu₇Te₄、Mo₆S₈、PTCDA 多类正极均具备普适提升效果,三层软包电芯能量密度达 134.5 Wh kg⁻¹,稳定循环 500 圈。依托分子动力学、原位电化学、冷冻透射、XPS 深度剖析完整解析氯锚定加速脱溶、双界面协同稳定微观机理,为无活化、长寿命镁离子电池提供通用电解液设计范式。①氯锚定添加剂创新:首次提出 EMM⁺阳离子氯离子捕获策略,从溶剂化根源弱化 Mg-Cl 强配位,彻底消除镁电池长活化短板;②动力学机理创新:区分溶剂鞘束缚强弱,证实离子液体调控可拉长 Mg-Cl 键、提升自由离子占比,系统建立脱溶能垒与电池活化周期定量关联;③双界面协同创新:添加剂同步优化镁负极沉积形貌与转化型正极 CEI 组分,生成薄型富MgCl₂无机界面,稳定 Cu₇S₄可逆中间产物;④体系普适创新:适配铜基硫 / 硒化物、钼硫、有机共轭多类正极,可放大至安时级软包电芯,具备规模化长时储能应用价值。图 1 不同离子液体阳离子氯锚定能力与电解液调控机理示意图(a) 多种离子液体阳离子对 Cl⁻吸附能 DFT 计算对比;(b) 纯 APC 电解液:强 Mg-Cl 配位、脱溶缓慢、需数十圈活化;(c) 中等锚定 APC-PP₁₄体系:活化周期缩短;(d) 强锚定 APC-EMM 体系:Mg-Cl 作用大幅减弱,无活化、均匀镁沉积1)EMM⁺对氯离子结合强度远高于哌啶类离子液体 PP₁₄⁺,可高效竞争溶剂鞘内氯离子;2)纯 APC 电解液 Mg-Cl 键长短、配位紧密,脱溶阻力大,镁沉积凹凸不平且伴随深度腐蚀;3)引入 EMM⁺后 Mg-Cl 键拉伸,溶剂鞘松散,镁离子脱溶动力学大幅提速,镁沉积致密无裂纹;4)氯锚定效应强弱直接对应电池活化圈数,锚定越强,达到满容量所需循环越少。(a-c) APC、APC-PP₁₄、APC-EMM 电解液分子动力学模拟快照;(d) 三类体系 Mg-Cl 径向分布函数 RDF;(e-g) 循环后镁负极表面与截面 SEM 形貌;(h) 塔菲尔极化曲线;(i-k) 镁对称电池 DRT 弛豫分布图谱;(l) 长循环极化曲线;(m) 已报道各类镁电解液循环时长对标1)RDF 结果显示 APC-EMM 体系 Mg-Cl 特征峰位置右移,键长显著拉长,氯离子从内层溶剂鞘剥离;2)纯 APC 负极循环后布满裂纹、腐蚀深度超 25 μm,APC-EMM 仅存在 4 μm薄腐蚀层,表面平整致密;3)APC-EMM 交换电流密度提升一个数量级,脱溶、SEI 形成对应阻抗大幅降低;4)Mg||Mg 对称电池稳定循环超 4000 h,循环寿命远超现有主流镁基电解液体系。图 3 CuS纳米片 || 镁全电池电化学性能对比(a) 0.1 A g⁻¹ 前三圈充放电曲线;(b) 循环伏安 CV 图谱;(c) 0.1 A g⁻¹ 长循环容量衰减曲线;(d) 梯度倍率充放电轮廓;(e) 倍率性能柱状图;(f) 近年镁电解液体系性能雷达对标;(g) 1 A g⁻¹ 千五百圈长效循环1)纯 APC 电解液需要 20 圈逐步爬升容量,APC-EMM 首圈即可达到最高容量,完全无活化阶段;2)CV 曲线氧化还原极化大幅降低,氧化峰由 2.0 V 降至 1.7 V,氧化还原可逆性显著提升;3)0.1 A g⁻¹ 初始容量 405.1 mAh g⁻¹,循环百圈容量保持 98.3%;1 A g⁻¹ 循环 1500 圈剩余 83.8% 容量;4)高低电流下均无明显极化抬升,倍率稳定性优于绝大多数文献报道镁电解液。(a) 不同放电深度原位 XRD 图谱;(b) 电解液内各类阳离子在CuS表面吸附模型;(c) DFT 吸附能定量对比;(d) Cu-S 二元相图;(e) CuS可逆转化完整反应机理1)放电过程CuS逐步转化为 Cu₇S₄中间相,深度还原生成金属 Cu,充电可完全可逆恢复原始物相;2)EMM⁺在CuS表面吸附能远低于 Mg²⁺、MgCl⁺,不会参与正极氧化还原,仅调控电解液本体溶剂化;3)EMM⁺可热力学稳定 Cu₇S₄中间产物,相比 APC 体系Cu₂S中间体,转化反应更完全,容量利用率更高;4)多谱学联合证实 EMM⁺仅作用于溶剂鞘,不嵌入正极晶格,无不可逆离子损耗。图 5 循环后正极 CEI 界面冷冻透射与ToF-SIMS 深度剖析(a) APC 电解液循环后CuS冷冻 TEM 图;(b) APC-EMM 体系界面形貌;(c-e) Cl⁻、Mg⁻、MgCl₃⁻二次离子质谱深度曲线;(f-h) 对应元素三维分布重构1)纯 APC 生成厚达 15 nm 富有机杂乱界面,APC-EMM 仅 5 nm 致密MgCl₂无机 CE 膜,厚度降低 66.7%;2)APC-EMM 体系界面无机氯化物占比大幅提升,惰性 MgO 副产物含量显著下降;3)ToF-SIMS 证明改性电解液体系电极内部游离氯、镁络合离子残留更少,不可逆副反应被抑制;4)薄型富MgCl₂界面可加速镁离子跨界面传输,循环过程阻抗增长极缓。(a-b) CuSe正极高低倍率循环;(c-d) PTCD 有机正极充放电与倍率;(e-f) Mo₆S₈钒基正极性能;(g) 四类正极 200 圈容量保持率对比;(h) 三层CuS镁软包长循环曲线;(i) 软包选取圈充放电轮廓;(j) 文献软包电池能量 / 寿命对标1)CuSe、Cu₇Te₄、Mo₆S₈、PTCDA 四类正极使用 APC-EMM 后活化圈数大幅缩短,容量、倍率同步提升;2)500 mA g⁻¹ 下三层软包电池循环 500 圈容量保留 92.7%,单体能量密度 134.5 Wh kg⁻¹;3)软包可稳定驱动 LED 模组,弯折后无漏液、无短路,具备储能工程实用潜力;4)同规格镁软包对比,本体系循环稳定性与能量密度处于行业领先水平。本研究借助 DFT 氯锚定阳离子筛选,开发 EMM⁺离子液体添加剂改性 APC-EMM 电解液。EMM⁺可强捕获电解液内氯离子,拉伸 Mg-Cl 配位键、打散致密溶剂鞘,从动力学层面大幅降低 Mg²⁺脱溶势垒,彻底消除镁电池漫长活化周期。电解液同时实现正负极双界面协同优化:镁金属侧诱导无腐蚀、平整致密沉积;转化型CuS正极原位生成薄型富MgCl₂无机 CEI,热力学稳定 Cu₇S₄可逆中间相,提升转化反应完整度。CuS||Mg 扣式电池 0.1 A g⁻¹ 容量 405.1 mAh g⁻¹,1 A g⁻¹ 循环 1500 圈容量保有 83.8%;该电解液对铜基硫硒化物、Mo₆S₈、有机 PTCDA 正极均具备普适提升效果,放大制备三层软包电芯能量密度可达 134.5 Wh kg⁻¹,稳定循环 500 圈。结合分子动力学模拟、原位电化学、冷冻透射、XPS/ToF-SIMS 多尺度表征完整阐明氯锚定加速脱溶、双界面长效稳定作用机理,为无活化、高可逆、可规模化镁离子储能电池提供通用电解液分子调控策略。High-Performance, Activation-Free Magnesium-Ion Batteries Enabled by Ionic Liquid Electrolyte Additive. Angew. Chem. Int. Ed., 2026; https://doi.org/10.1002/anie.7580996本文内容来源于学术研究论文,版权归原作者所有。转载旨在分享学术成果,仅供参考,不构成任何应用建议。如涉及作品内容、版权或其他问题,请及时联系处理。



 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
