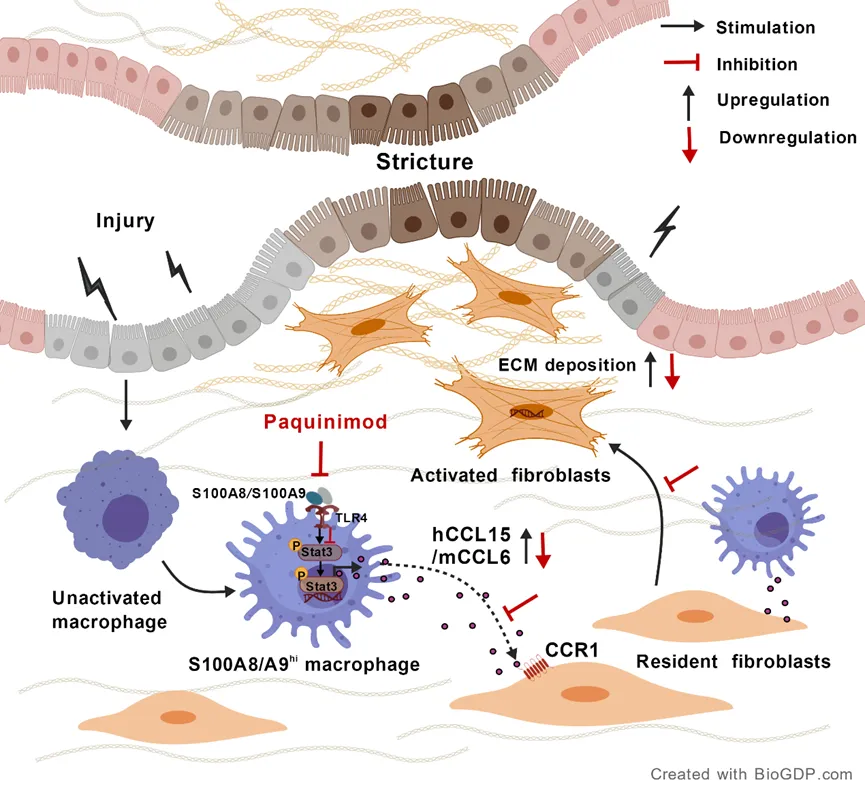
背景
克罗恩病(CD)肠道纤维化主要由炎症或损伤刺激下活化的成纤维细胞驱动,导致细胞外基质(ECM)成分的过度积累,大量的纤维组织沉积并取代正常实质组织,最终引起肠腔进行性狭窄。尽管CD相关肠纤维化的特征研究已取得进展,但肠成纤维细胞活化和纤维化进展的具体分子驱动因素仍待进一步阐明。
除成纤维细胞自身的效应外,免疫微环境——特别是调控组织修复的调控网络,在启动和维持纤维化级联反应中亦发挥关键作用。巨噬细胞是维持组织稳态的免疫哨兵,其具有高度的异质性和显著的表型可塑性。在稳态下,巨噬细胞作为修复性哨兵发挥作用;而在免疫失调后,则转变为促纤维化的驱动者。尽管巨噬细胞在各种器官纤维化中的促纤维化作用已得到充分证实,但其在CD相关肠道纤维化中的作用在很大程度上仍未被探索。
在该研究中,研究者们首先通过重新分析CD全层肠组织的单细胞测序数据,鉴定出一个独特的S100A8/A9高表达(S100A8/A9hi)巨噬细胞亚群,并证实其在肠道狭窄区域的显著浸润。为确定其在肠纤维化中的作用,研究者在葡聚糖硫酸钠(DSS)诱导的结肠炎小鼠模型中,采用了骨髓来源的S100A8/A9hi巨噬细胞过继转移。接着研究者探究了靶向S100A8/A9在慢性结肠炎动物模型中的治疗效果。为进一步鉴定该亚群巨噬细胞中关键的促纤维化介质,其进行了蛋白质组学测序并建立了共培养体系。机制上,研究者阐明了S100A8/A9hi巨噬细胞如何介导小鼠CCL6(mCCL6)的产生。最后,研究者证实mCCL6的人类同源物CCL15(hCCL15)可通过CC趋化因子受体1(CCR1)调控肠成纤维细胞,进而发挥促纤维化作用。
方法
研究者重新分析CD纤维性狭窄肠组织全层单细胞测序数据,确定待研究的目的细胞亚群。收集CD患者狭窄及非狭窄部位肠道组织手术标本,分别采用免疫组化及WB检测S100A8/A9的表达,利用免疫荧光检测S100A8/A9+巨噬细胞在CD狭窄部位肠组织中的浸润及其与成纤维细胞的空间位置毗邻关系。提取并诱导小鼠骨髓源巨噬细胞(BMDM),利用脂多糖(LPS)诱导S100A8/A9hi巨噬细胞,或利用S100a9的小干扰RNA进行反向验证。在野生型小鼠体内构建DSS诱导的慢性结肠炎模型,分别将NC-BMDM,S100A8/A9hi-BMDM以及siNC-BMDM与siS100a9-BMDM分批次过继移植至小鼠体内进行正反验证,活体成像进行体内示踪,造模结束后评估小鼠肠道炎症及肠纤维化水平。利用靶向S100A8/A9的抑制剂——帕奎莫德(Paquinimod, PAQ)灌胃治疗,评估小鼠肠道纤维化程度。体外实验中,分别将NC-BMDM,S100A8/A9hi-BMDM与人原代肠成纤维细胞(HIF)共培养,检测其对HIF活化、增殖和ECM生成的影响。收集siNC-BMDM与siS100a9-BMDM培养上清行蛋白质组学检测以筛选差异蛋白分子,并在体内外探究该分子对结肠炎小鼠肠道纤维化以及HIF纤维化表型的影响。
结果
1. S100A8/A9hi巨噬细胞在CD患者纤维性狭窄肠段中浸润增加
为探究肠道纤维化中的免疫细胞动态,研究者分析了CD患者的全层肠组织单细胞测序数据集,包括非受累区、炎症区(非狭窄)和狭窄区。基于既往研究已发现狭窄区域成纤维细胞-髓系细胞亚群间存在强烈的细胞间通讯信号,因此研究者聚焦于髓系细胞亚群。首先通过无监督聚类将髓系细胞群注释为15个亚群(图S1A、B和图1A),随后采用计算工具scDist量化这些亚群在三组中的细胞间距离。结果发现,其在炎症区vs非受累区以及狭窄区vs炎症区的比较中,S100A8/A9hi巨噬细胞的细胞距离评分均位居首位(图1B),并在狭窄部位的占比显著增加(图1C)。细胞间互作网络分析进一步提示S100A8/A9hi巨噬细胞亚群与成纤维细胞之间存在强烈的细胞间通讯信号(图1D),这部分结果凸显了S100A8/A9hi巨噬细胞的异质性,提示其可能是参与CD肠纤维化的关键驱动因素。
接下来,研究者进行了组织病理学验证。对CD肠段进行的H&E和Masson染色证实了狭窄区域存在显著的黏膜下层扩张和胶原沉积。免疫组化和WB分析均显示,相比非受累区,狭窄区的S100A8/A9表达水平更高(图1E、F)。免疫荧光分析不仅证实了S100A8/A9+巨噬细胞在CD狭窄肠组织中的富集浸润(图1G),还揭示了其与活化的成纤维细胞间存在密切的空间毗邻关系(图1H)。与此同时,在DSS慢性结肠炎小鼠的结肠组织中也观察到了类似的显著浸润(图1I),强调了此为纤维化的一个共性特征。综上所述,这些发现将S100A8/A9hi巨噬细胞鉴定为CD肠纤维化发病中的关键免疫细胞亚群,并将其作为进一步的研究焦点。
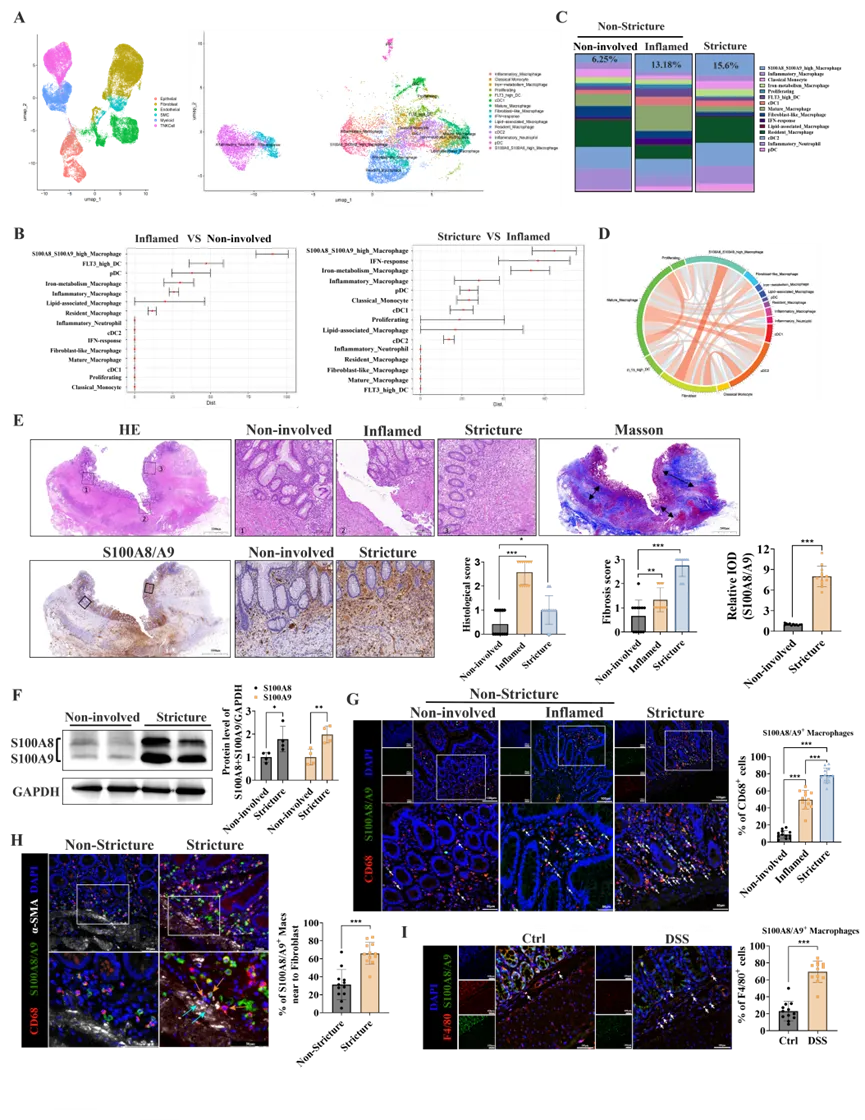
图1. S100A8/A9hi巨噬细胞在CD肠维化区域大量浸润
2. S100A8/A9hi巨噬细胞加重DSS诱导的慢性结肠炎小鼠肠道纤维化
为探究S100A8/A9hi巨噬细胞在肠道纤维化中的作用,研究者在2% DSS诱导的结肠炎小鼠中建立了过继转移模型。体外利用LPS刺激BMDMs以获得S100A8/A9hi巨噬细胞。在过继移植前给予氯膦酸盐脂质体以清除小鼠内源性巨噬细胞,自结肠炎诱导后第7天起,小鼠每周接受一次尾静脉输注PBS、正常对照BMDMs(NC-BMDMs)或S100A8/A9hi BMDMs(图2A),通过活体成像证实了回输的BMDMs成功在结肠组织中归巢(图2B)。
在肠道炎症水平上,与NC-BMDMs组相比,S100A8/A9hi BMDMs的过继转移显著加重了结肠炎:表现为体重下降加剧(图2C)、结肠长度缩短(图2D)、炎症因子水平升高(图2E)以及组织病理学损伤加重(图2F)。肠道纤维化程度上,S100A8/A9hi BMDMs的过继转移显著加重了结肠炎小鼠肠道纤维化,表现为胶原沉积增加(图2G)、I型胶原阳性肌层增厚(图2H)以及纤维化相关基因表达上调(图2I)。此部分结果证实了S100A8/A9hi巨噬细胞在结肠炎相关肠道纤维化发病中的强促纤维化作用。
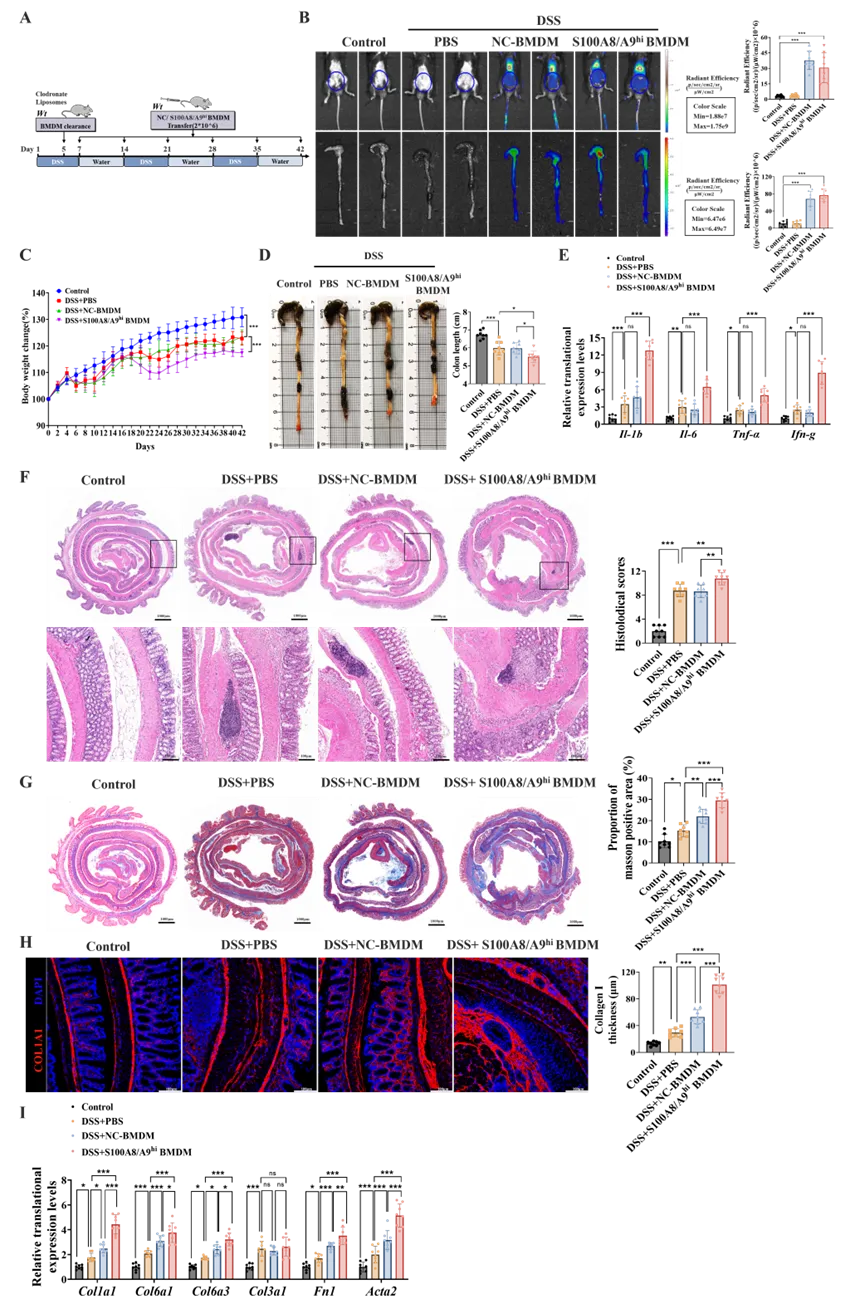
图2. S100A8/A9hi巨噬细胞过继转移加重DSS慢性结肠炎小鼠肠道纤维化
3. S100A8/A9缺陷的巨噬细胞促纤维化能力减弱
为进一步验证上述发现,研究者利用靶向S100a9的小干扰RNA转染BMDMs,从而导致巨噬细胞中S100A8/A9蛋白复合物缺陷。接着将siNC-BMDMs以及siS100a9-BMDMs回输至结肠炎小鼠体内。活体成像证实转移后1、6、24和48小时受体小鼠的腹部荧光信号增强(图S3A);结肠组织切片的荧光信号证实了过继移植的巨噬细胞在结肠组织中的成功归巢(图S3D)。
造模结束后,研究者评估了各组小鼠的肠道炎症严重程度:与siNC-BMDMs过继转移组相比,接受S100A8/A9缺陷BMDMs的小鼠炎症表型显著改善,表现为体重更高(图3A)、炎症因子水平更低(图3B)、结肠长度更长(图3C)以及病理损伤减轻(图3D)。随后,研究者评估了各组小鼠的肠道纤维化水平。与接受siNC-BMDMs的小鼠相比,siS100a9-BMDMs处理组的慢性结肠炎小鼠肠道纤维化显著减轻,表现为胶原、纤连蛋白及α-SMA表达的明显下调(图3E、F)。组织学分析进一步显示,该组小鼠胶原沉积显著减少(图3G),且α-SMA阳性肌层增厚程度明显降低(图3H)。综上所述,这些结果表明高表达S100A8/A9的巨噬细胞是驱动结肠炎相关肠道纤维化的关键因素。
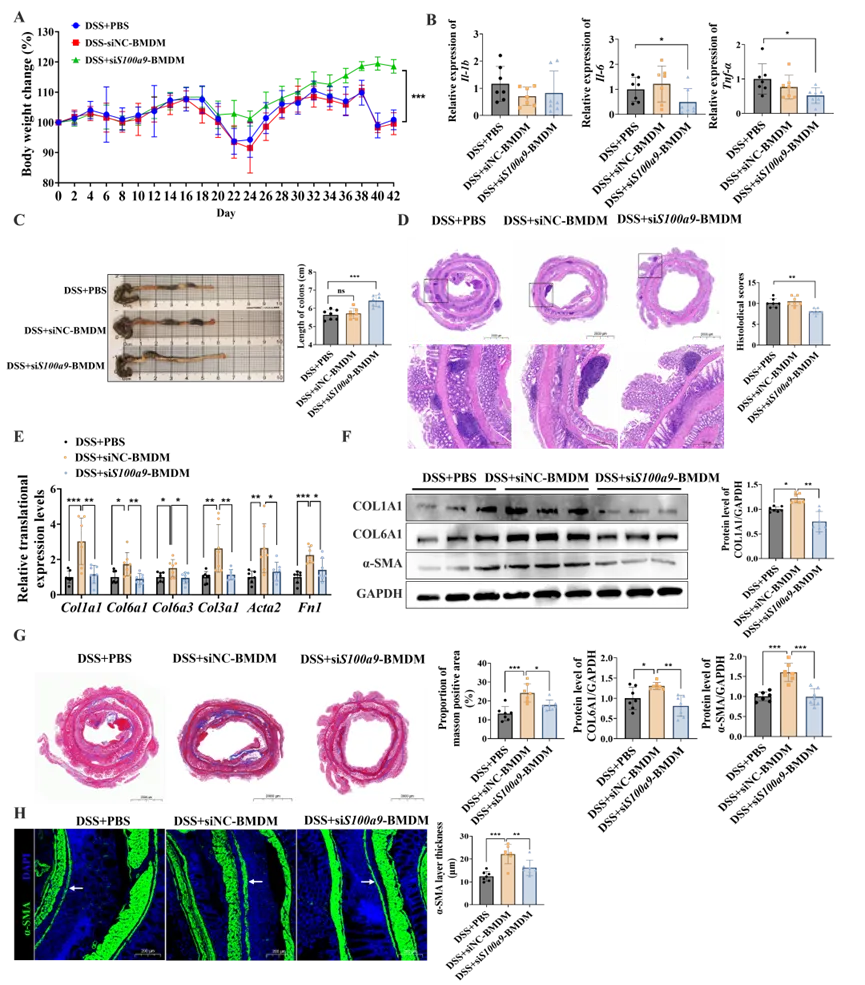
图3. S100A8/A9缺陷的巨噬细胞在慢性结肠炎小鼠中表现出促纤维化能力受损
4.靶向S100A8/A9减轻慢性结肠炎小鼠肠道纤维化
鉴于S100A8/A9hi巨噬细胞在肠道纤维化发病中的关键作用,研究者进一步评估了药理学抑制S100A8/A9的治疗潜力。研究者自第二周期的DSS诱导开始每日灌胃给予1、5或10 mg/kg/d 的S100A8/A9抑制剂——PAQ(图4A)。治疗结束后,首先通过免疫荧光和流式细胞术检测各组小鼠结肠组织中S100A8/A9+巨噬细胞的浸润情况,结果显示DSS诱导后该细胞群显著扩增,而10 mg/kg/d剂量的PAQ治疗显著减少了该亚群的浸润(图4B、C)。相应地,该剂量的PAQ显著改善了结肠炎小鼠的炎症程度,具体表现为体重下降和结肠缩短的缓解(图S4A、B)、炎症因子表达水平的抑制(图S4C)以及组织学损伤的减轻(图4D)。肠道纤维化程度的评估发现,三种剂量的PAQ均显著改善了肠道纤维化,且高剂量组(10 mg/kg/d)效果最为突出,表现为肠组织中胶原沉积减少(图4E)、α-SMA阳性肌层厚度减少(图4F)以及ECM成分(包括I型胶原、VI型胶原和纤连蛋白)表达下调(图4G、H)。
为评估PAQ的安全性,研究者监测了各组小鼠的血液学指标。PAQ整体耐受性良好,研究期间未观察到药物相关死亡。10 mg/kg/d剂量的PAQ虽引起单核细胞、淋巴细胞和中性粒细胞计数及转氨酶水平的轻度变化,但所有指标均处于正常生理范围内(表S4),提示PAQ具有良好的安全性。这些数据表明靶向S100A8/A9可能是一种安全有效的改善结肠炎相关肠道纤维化的策略。
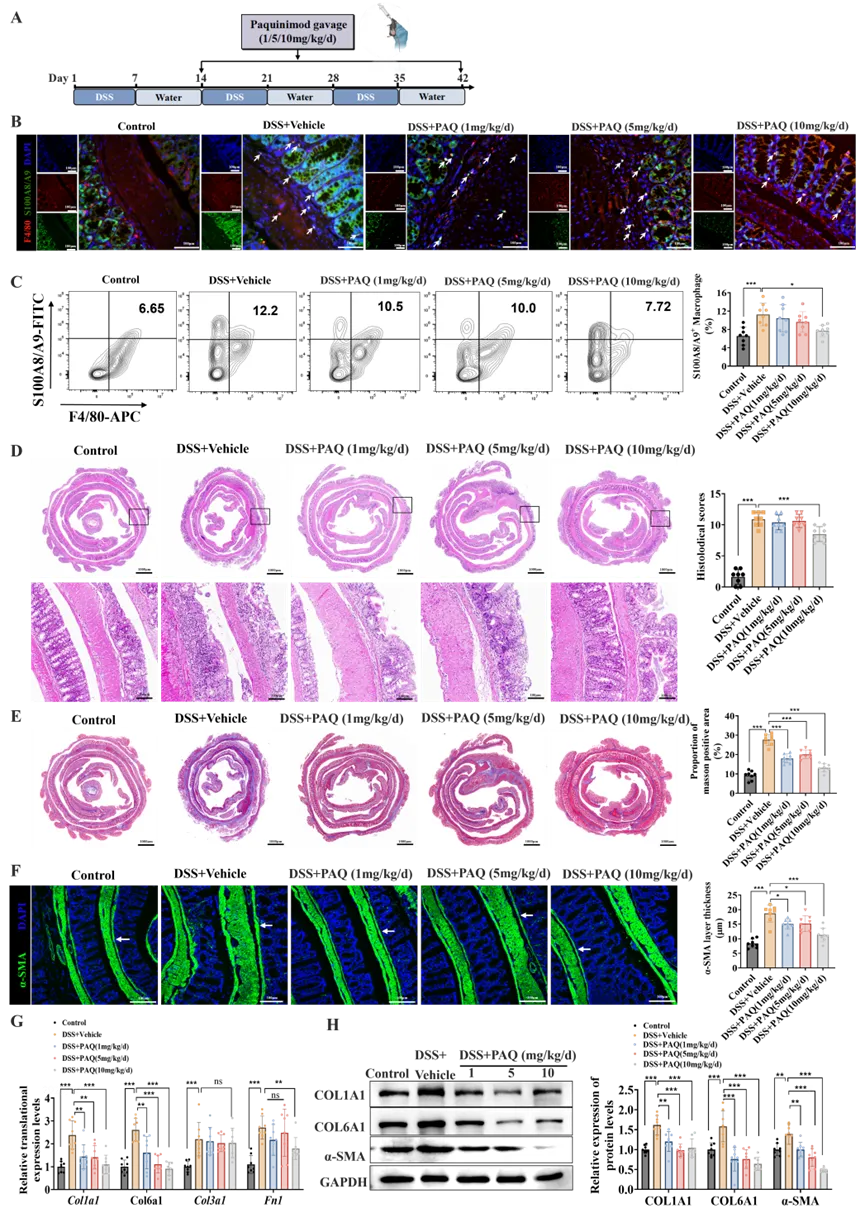
图4. S100A8/A9作为缓解DSS结肠炎小鼠肠道纤维化的有效治疗靶点
5. S100A8/A9hi巨噬细胞促进肠成纤维细胞的活化与ECM产生
为探究S100A8/A9hi巨噬细胞促进肠纤维化的机制,研究者建立了小鼠胚胎成纤维细胞(NIH-3T3)与BMDMs体外共培养体系(图5A)。EdU增殖实验显示,与NC-BMDMs共培养相比,S100A8/A9hi BMDMs共培养显著促进了成纤维细胞增殖(图5B)、诱导成纤维细胞活化,并促进ECM关键成分的产生(图5C、D)。反之,BMDMs中S100a9的敲低显著削弱了其促纤维化能力。具体而言,相较于siNC-BMDMs,与siS100a9-BMDMs共培养时,成纤维细胞增殖明显降低(图5E、F),且伴随着成纤维细胞活化的减弱以及纤连蛋白和多种胶原表达水平的显著下调(图5G、H)。结果表明S100A8/A9hi巨噬细胞通过直接驱动成纤维细胞增殖、活化和ECM合成来促进肠道纤维化。
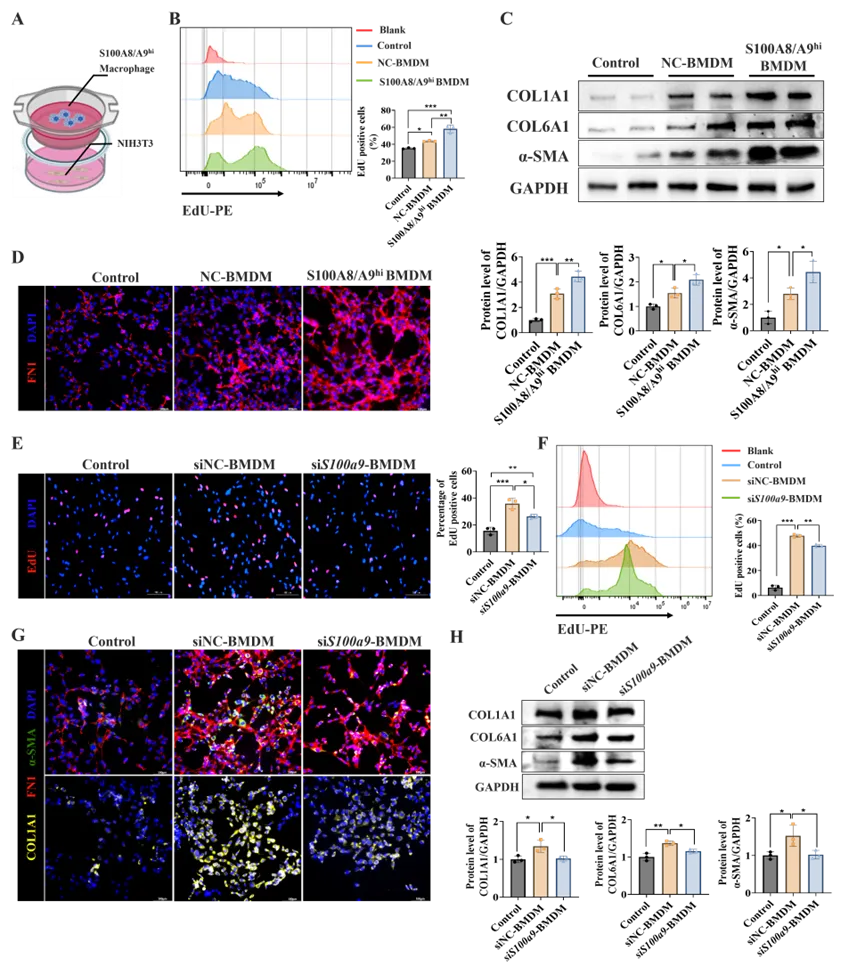
图5. S100A8/A9hi巨噬细胞驱动肠成纤维细胞增殖、活化和ECM生成
6. S100A8/A9hi巨噬细胞通过分泌趋化因子CCL6参与加剧肠道纤维化
为鉴定S100A8/A9hi巨噬细胞分泌的关键促纤维化介质,研究者收集了对照组和S100a9敲低后BMDMs的条件培养基进行蛋白质组学分析,共鉴定出64个差异表达蛋白(23个上调,41个下调;图S5A)。KEGG分析显示差异分子显著富集于与纤维化密切相关的信号通路—ECM-受体相互作用通路(图6A)。在变化最显著的差异分子中,研究者基于其分泌特性及其在其他器官纤维化中的已知作用,优先选择趋化因子mCCL6进行深入研究(图6B、C)。
为探究mCCL6的功能作用,研究者在接受S100A8/A9hi BMDMs移植的DSS结肠炎小鼠体内给予CCL6中和抗体。抗体治疗有效抑制了小鼠结肠组织和血清中mCCL6水平(图S5B、C),并显著缓解肠道炎症,表现为体重改善(图S5D)、结肠缩短减轻(图S5E)、炎症因子表达降低(图S5F)及病理损伤改善(图6D)。mCCL6的阻断还显著改善了肠道纤维化,表现为肠组织中胶原沉积减少(图6E)、I型胶原阳性肌层厚度减少(图6F)、纤维化相关基因(Col1a1、Col6a1、Col6a3和Col3a1)(图S5G)以及COL6A1和α-SMA蛋白水平降低(图6G)。
体外实验进一步支持上述发现:共培养体系中加入抗CCL6中和抗体后,S100A8/A9hi BMDMs对成纤维细胞的增殖(图S6A、B)、活化及ECM产生的促进能力均减弱(图S6C、D)。此外,CCL6受体CCR1的特异性抑制剂显著削弱了S100A8/A9hi BMDMs诱导的成纤维细胞增殖、ECM生成(图S7D、E)、迁移以及收缩能力(图S7F-G)。这些发现表明mCCL6是S100A8/A9hi巨噬细胞驱动结肠炎相关纤维化的关键下游介质,且主要通过CCR1信号通路发挥作用。
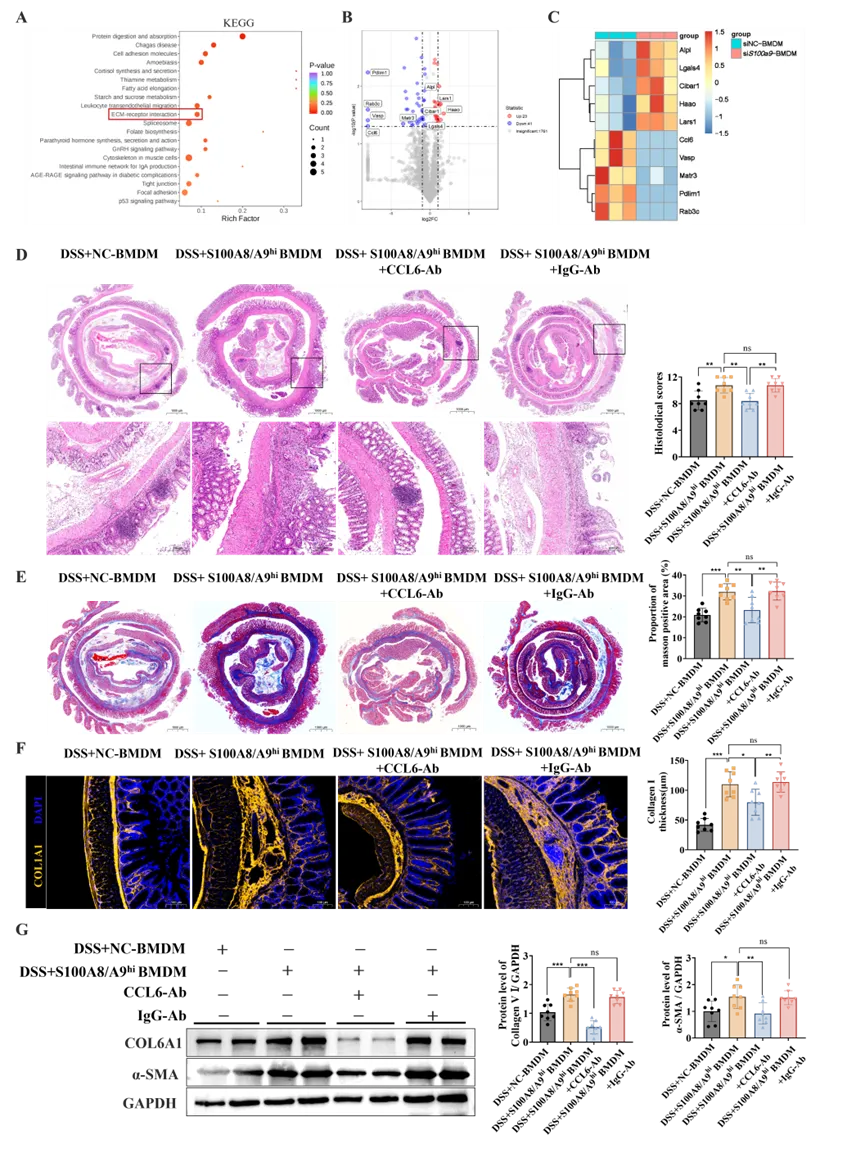
图6. S100A8/A9hi巨噬细胞通过mCCL6配体驱动结肠炎相关的肠道纤维化
7. S100A8/A9hi巨噬细胞通过激活STAT3介导CCL6生成
为探究S100A8/A9hi巨噬细胞中CCL6产生的调控机制,研究者利用JASPAR、TFBs和KnockTF三个数据库进行整合交集分析,鉴定出9个参与调控CCL6的潜在转录因子(Atoh1、Bcl6、Elf5、Meis1、Myb、Myc、Sox11、Stat3和Stat4)(图7A)。STRING蛋白-蛋白相互作用分析显示,S100A9与STAT3之间存在强且特异性的关联(图7B)。鉴于S100A9在其他病理背景下调控STAT3的已知作用,本研究进一步验证了这一关系:重组S100A8-S100A9蛋白刺激后,STAT3磷酸化显著增加(图7C)。随后,研究者通过特异性抑制剂检测S100A8/A9是否经由TLR4和RAGE这两条通路激活STAT3。结果显示,TLR4抑制剂显著减弱了p-STAT3表达和CCL6分泌,而RAGE抑制无明显影响(图7D、E);且TLR4抑制后,S100A8/A9与TLR4的共定位减少(图7F),证明了S100A8/A9主要通过TLR4信号诱导下游STAT3激活。
接下来,研究者检测了STAT3激活是否诱导CCL6产生。STAT3抑制剂Stattic显著减弱了S100A8-S100A9刺激后巨噬细胞的CCL6分泌(图7G、H)。为在内源性条件下验证上述发现,siRNA介导的S100a9敲低同时减少了下游STAT3磷酸化和CCL6蛋白水平;而STAT3激活剂Colivelin TFA有效挽救了S100a9敲低导致的CCL6下调(图7I)。为进一步确认S100A9-STAT3轴,研究者进行了核质分离实验,结果显示S100a9敲低显著减少了BMDMs胞质和胞核中的STAT3磷酸化水平,且该抑制作用可被STAT3激活剂有效逆转(图7J、K)。最后,为确认STAT3是否直接调控CCL6转录,基于JASPAR数据库预测,研究者在Ccl6启动子中鉴定出四个经典STAT3结合基序(图7L)。双荧光素酶报告基因实验显示,STAT3显著增强了野生型Ccl6启动子的转录活性,而结合位点突变后该效应减弱(图7M)。此外,ChIP实验证实S100A8-S100A9刺激后STAT3与Ccl6启动子的结合显著增强(图7N、O)。此部分结果表明S100A8/A9hi BMDMs主要通过激活STAT3驱动CCL6的生成。
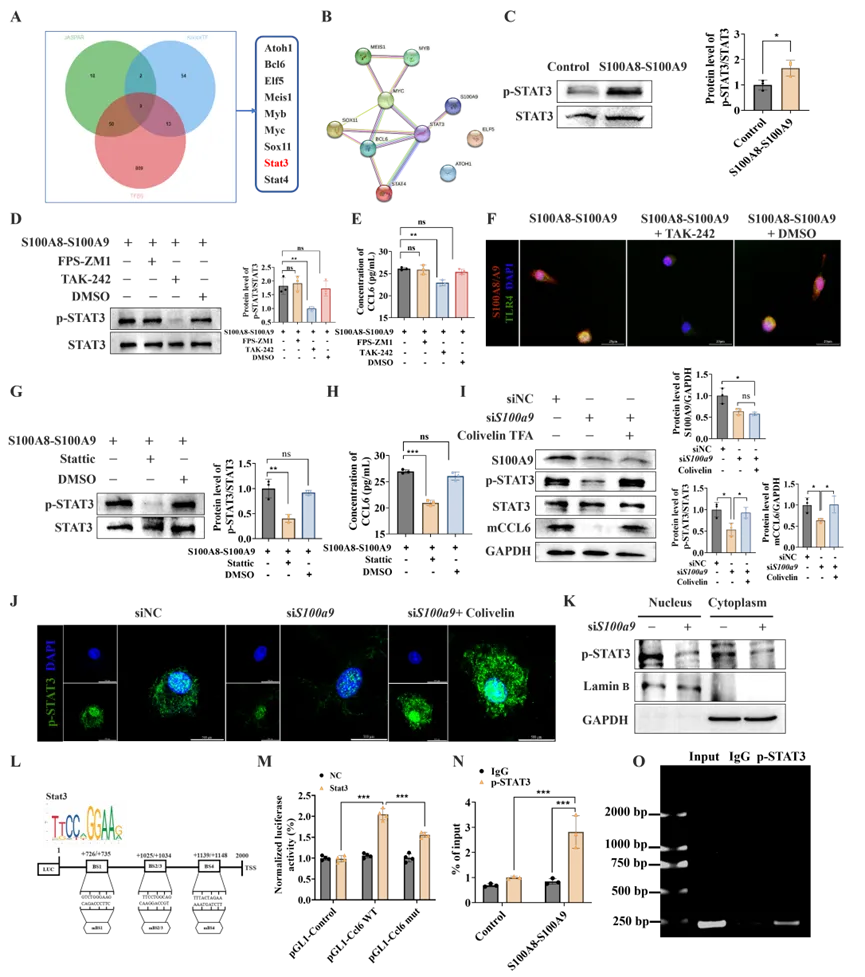
图7. S100A8/A9hi巨噬细胞通过激活转录因子STAT3介导CCL6表达
8. 人类同源基因hCCL15通过CCR1对成纤维细胞发挥类似的促纤维化作用
小鼠CCL6的功能性人类同源物为hCCL15和hCCL23。人类蛋白质图谱数据显示,hCCL15在肠道中高表达,而hCCL23呈低水平非特异性表达(图S8A)。临床样本验证发现,hCCL15在狭窄区域高表达并与胶原沉积共定位(图8A、B),而CCL23无显著差异(图S8B、C)。此外,在伴有肠道狭窄的CD患者血清中hCCL15水平亦显著高于无狭窄患者(图8C),且转录组分析显示CCL15表达与纤维化相关基因,如COL1A1、COL3A1和FN1呈正相关。因此研究者后续研究聚焦于hCCL15。
为评估hCCL15的直接促纤维化效应,研究者从CD患者肠道狭窄区域分离HIFs,分别利用不同浓度的hCCL15重组蛋白(0、5、10和20 ng/mL)处理,结果发现10和20 ng/mL的hCCL15均可显著促进COL1A1合成,但更高剂量无进一步增加(图8E),故选择10 ng/mL进行后续实验。接下来的功能验证发现,hCCL15可显著促进HIFs迁移(图8F),并增强其活化与增殖能力(图8G);同时显著上调ACTA2、COL1A1、COL3A1、COL6A1、COL6A3和FN1等相关纤维化基因的转录水平(图8H)。
由于hCCL15主要通过下游受体CCR1和CCR3发挥功能,研究者分析了CD全层肠组织scRNA-seq数据,发现成纤维细胞亚群中CCR1表达水平显著高于CCR3(图S8D),并在配对的非狭窄/狭窄肠组织以及相应部位提取的原代成纤维细胞中得到了验证(图S8E、F)。多重免疫荧光进一步证实纤维化狭窄肠组织中S100A8/A9+ CCL15+巨噬细胞与CCR1+ α-SMA+成纤维细胞空间相邻(图8I),提示狭窄区域存在CCL15-CCR1信号传导。为验证CCR1的功能作用,研究者应用了 CCR1拮抗剂BX471,发现 CCR1阻断显著减弱了hCCL15诱导的HIFs的胶原产生(图S8G)、细胞迁移(图S8H)及其增殖和活化能力(图S8I)。这些结果证明,人类同源物hCCL15通过CCR1直接参与成纤维细胞的活化、增殖、迁移和胶原合成能力,从而在CD中参与驱动肠道纤维化进展。
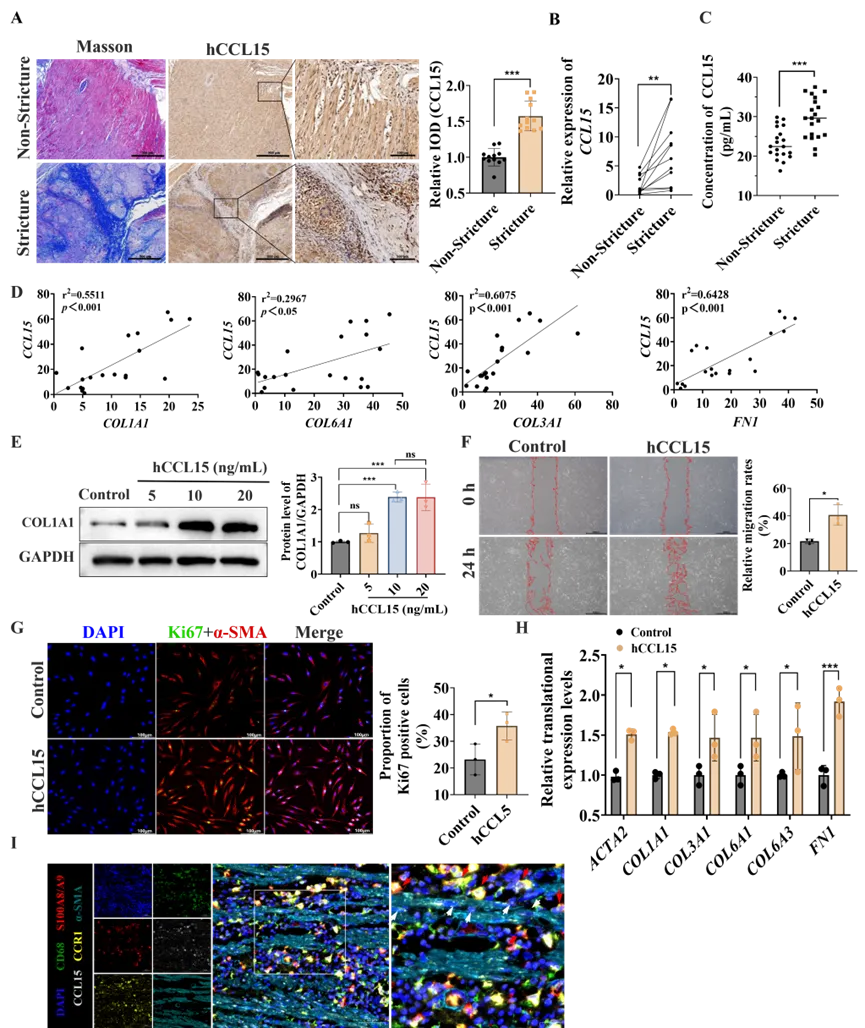
图8. hCCL15促进人肠成纤维细胞的活化、增殖、迁移以及ECM生成
结论
该研究证实了S100A8/A9高表达巨噬细胞是CD肠道纤维化狭窄形成中的关键致病亚群。药理学靶向S100A8/A9可显著缓解结肠炎小鼠肠道纤维化。机制上,S100A8/A9hi巨噬细胞通过激活STAT3驱动mCCL6生成,后者经成纤维细胞CCR1信号通路发挥主要促纤维化介质作用。此外,其人类同源物hCCL15具有类似的促纤维化效应。通过将S100A8/A9、STAT3、mCCL6/hCCL15及CCR1确定为潜在治疗靶点,该研究为CD肠道纤维化的靶向干预提供了新的治疗策略。


