南京医科大学金陵医院 | 揭秘 DN 发病新机制:UHRF1 双调控 GRP78 失衡,核转运抑制剂带来肾脏保护新希望
- 2026-06-08 22:53:55

导读
为阐明糖尿病肾病(DN)中肾小管上皮细胞损伤的内质网(ER)应激调控机制,研究以高糖环境下 UHRF1 下调为切入点,通过细胞、动物及临床样本验证,发现 UHRF1 可通过启动子甲基化和 K48 连接泛素化双重调控 GRP78 稳态,其下调导致 GRP78 胞质蓄积并经 importin β1 介导核转运,核内 GRP78 激活 ER 应激及凋亡相关基因转录,加剧细胞损伤;经高通量筛选获得 GRP78 核转运抑制剂 Parishin,其可结合 GRP78 核定位信号域阻断核移位,在保留 GRP78 胞质分子伴侣功能的同时减轻 ER 应激与 DN 病理损伤,为 DN 提供了新型治疗靶点与策略。
注:该文章发表于《Kidney International》,该期刊最新影响因子为12.6,CiteScore达21.40,位列JCR泌尿学与肾脏学Q1区。
研究亮点
首次发现表观调控因子 UHRF1 对 GRP78 的双重表观调控机制(启动子甲基化 + 蛋白泛素化);明确 GRP78 经 importin β1 介导核转运并作为转录调控因子激活 ER 应激 / 凋亡通路;通过双平台筛选获得特异性 GRP78 核转运抑制剂 Parishin,其在保留 GRP78 生理功能的同时阻断病理作用,为 DN 提供了精准靶向治疗新策略。
研究背景
糖尿病肾病(DN)是糖尿病患者常见并发症,约 20%-40% 糖尿病患者会患病,且是终末期肾病的主要诱因,肾小管上皮细胞(RTEC)损伤在 DN 进展中起关键作用。代谢应激导致内质网(ER)稳态失衡,引发 ER 应激介导的细胞凋亡,是 DN 发病的重要病理机制,临床研究也证实 ER 应激标志物与 DN 患者蛋白尿及肾小管损伤相关。GRP78 作为核心 ER 分子伴侣,对维持 ER 稳态至关重要,高糖环境下其表达异常上调,但调控机制及功能作用尚未完全明确。此外,GRP78 可在应激下转运至胞外或细胞器发挥非经典功能,但其在 DN 中是否发生核转运及生物学意义仍不清楚,亟需揭示相关调控网络,为 DN 治疗提供新靶点。
研究方法
样本来源:收集 DN 患者与健康对照肾活检组织、db/db 自发糖尿病肾病小鼠及 db/m 对照小鼠肾组织;
细胞模型:采用人肾小管上皮细胞(HK-2)、大鼠肾小管上皮细胞(NRK-52E),经高糖(HG)持续培养构建细胞损伤模型;
分子生物学技术:通过 DNA pull-down、Co-IP、GST 下拉验证蛋白相互作用;采用甲基化特异性 PCR、Bisulfite 测序分析启动子甲基化;利用 Western blot、RT-qPCR 检测基因表达;通过 CUT&Tag 鉴定 GRP78 靶基因;
筛选与验证:结合分子对接与深度学习的双平台策略,从 210 万化合物库中筛选 GRP78 核转运抑制剂,经细胞毒性、SPR 结合实验及功能验证;
动物实验:db/db 小鼠经 Parishin 灌胃处理,检测肾功能指标、病理改变及相关分子表达;
统计学分析:采用 GraphPad Prism 9 进行 t 检验、单因素 / 双因素方差分析。
研究结果
高糖环境下 HK-2 细胞 GRP78 显著上调,伴随 ER 应激通路激活及细胞凋亡增加,DN 患者及 db/db 小鼠肾组织中 GRP78 表达随病情进展升高;
UHRF1 可结合 GRP78 启动子抑制其转录(依赖 H3K9me2/3 结合及半甲基化 CpG 结合活性),并通过与 GRP78 底物结合域(SBD)相互作用介导其 K48 连接泛素化降解,高糖诱导 UHRF1 下调导致 GRP78 蓄积;
GRP78 依赖核定位信号(NLS)与 importin β1 结合进入细胞核,核内 GRP78 结合 ER 应激 / 凋亡相关基因(ATF6、XBP1 等)启动子,激活转录;
高通量筛选获得抑制剂 Parishin,其与 GRP78 NLS 域高亲和力结合,阻断核转运,且不影响 GRP78 胞质分子伴侣功能;
Parishin 处理可显著降低 db/db 小鼠肾功能指标(血尿素氮、尿白蛋白 / 肌酐比等),减轻肾小球肥大、肾小管萎缩等病理改变,抑制 ER 应激与凋亡。
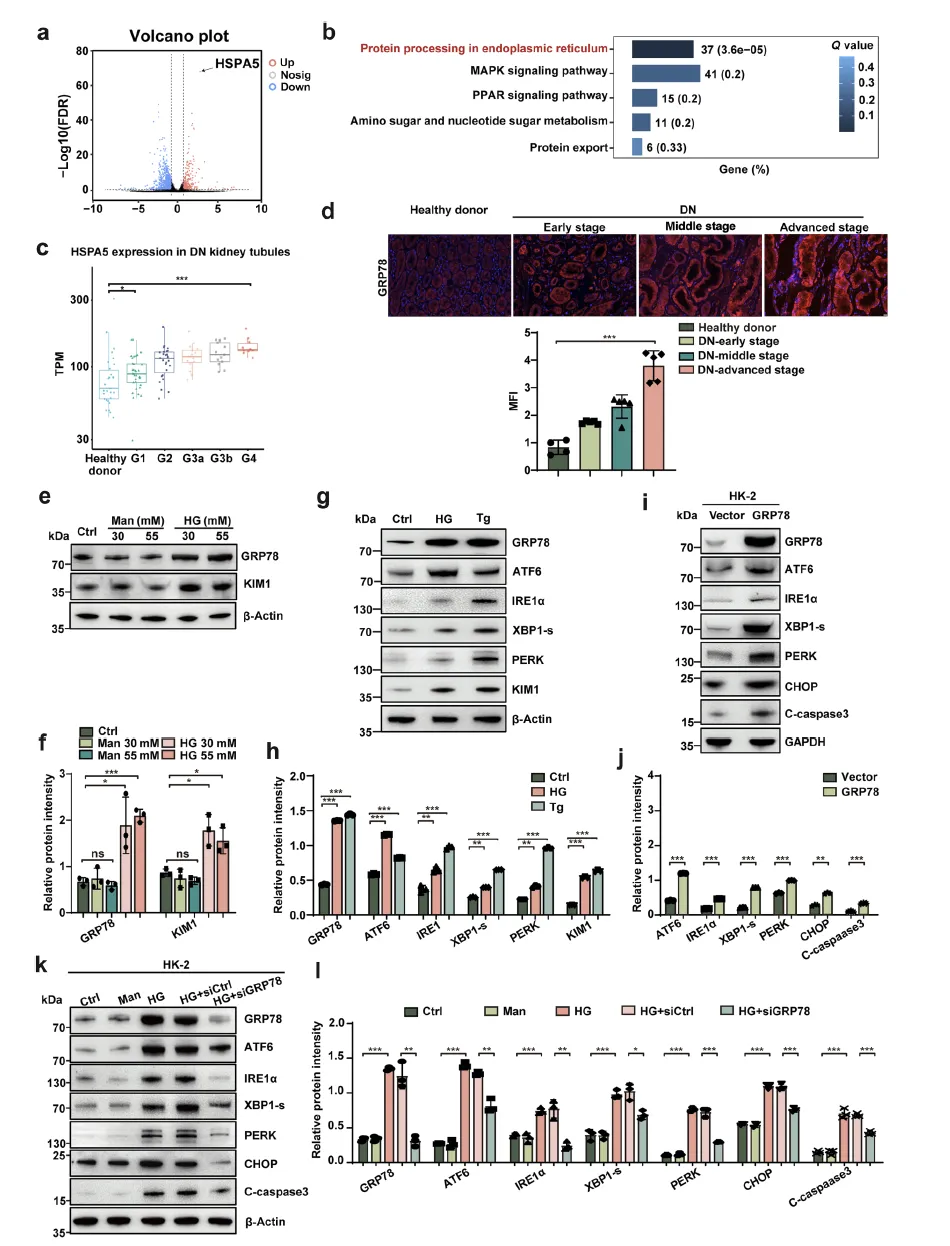
Figure 1:高糖诱导肾小管细胞 GRP78 上调加剧内质网应激
该图通过多维度实验验证了高糖环境下 GRP78 与糖尿病肾病(DN)中内质网(ER)应激的关联。首先,火山图分析显示高糖持续培养 10 代的 HK-2 细胞中,ER 应激相关基因 HSPA5(编码 GRP78)显著上调,KEGG 通路富集分析证实差异基因主要集中在 ER 蛋白加工、未折叠蛋白反应等通路。其次,临床样本数据表明,DN 患者肾小管中 HSPA5 mRNA 和 GRP78 蛋白表达随疾病分期(G1 至 G4)和病理损伤程度逐渐升高。细胞实验进一步显示,高糖(30/55 mM)处理可诱导 HK-2 细胞 GRP78 和损伤标志物 KIM1 表达增加,且 ER 应激诱导剂毒胡萝卜素能进一步放大 GRP78 水平。功能验证中,GRP78 过表达可激活 ATF6、IRE1α、XBP1-s 等 ER 应激通路关键分子及 CHOP、裂解型 caspase3 等凋亡标志物,而 GRP78 敲低则能减轻高糖诱导的 ER 应激和凋亡,明确 GRP78 积累是驱动 DN 肾小管损伤的病理性事件。
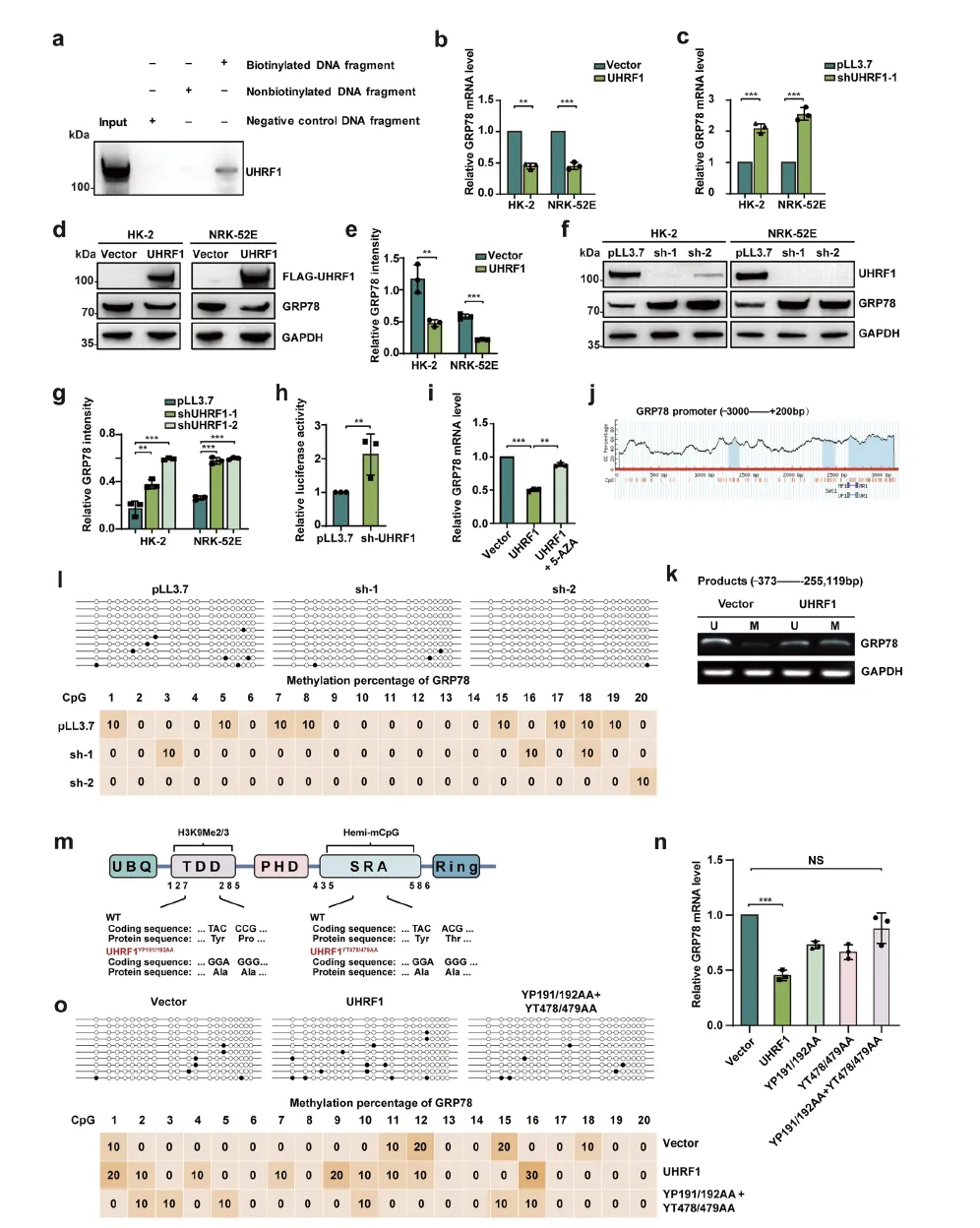
Figure 2:UHRF1 通过调控 GRP78 启动子甲基化调节其表达
该图聚焦 UHRF1 对 GRP78 的转录水平调控机制。DNA pull-down 结合质谱和 Western blot 证实,UHRF1 可直接结合 GRP78 启动子区域(–755 至 10 bp)。功能实验显示,UHRF1 过表达能显著降低 HK-2 和 NRK-52E 细胞中 GRP78 的 mRNA 和蛋白水平,而 UHRF1 敲低则促进 GRP78 表达,且 UHRF1 可抑制 GRP78 启动子的荧光素酶活性。DNA 去甲基化药物 5 - 氮杂胞苷处理后,UHRF1 对 GRP78 的抑制作用减弱,表明其调控依赖甲基化机制。甲基化特异性 PCR 和亚硫酸氢盐测序结果显示,UHRF1 过表达可增加 GRP78 启动子甲基化水平,而 DN 患者和 db/db 小鼠肾组织中 GRP78 启动子呈低甲基化状态。进一步构建 UHRF1 突变体发现,其对 GRP78 启动子甲基化的调控需要同时具备 H3K9me2/3 识别活性(YP191/192 位点)和半甲基化 CpG 结合活性(YT478/479 位点),缺失任一活性均会部分削弱调控效果,双重突变则完全丧失调控功能。
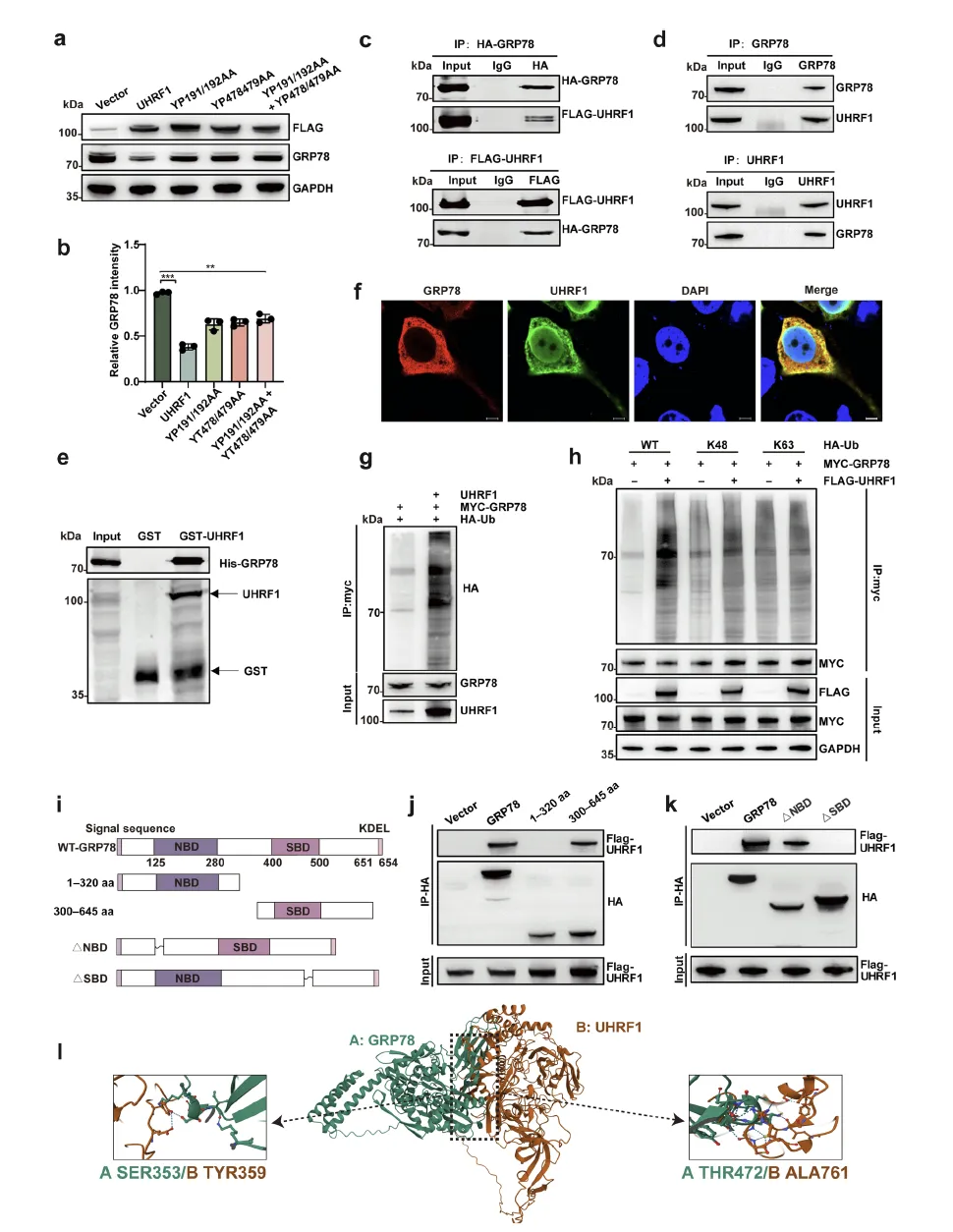
Figure 3:UHRF1 通过与 GRP78 的 SBD 结构域相互作用介导其 K48 连接泛素化
该图揭示了 UHRF1 对 GRP78 的翻译后调控机制。尽管 UHRF1 双重突变体(YP191/192AA+YT478/479AA)丧失了甲基化调控能力,但仍能抑制 GRP78 蛋白水平,提示存在额外调控方式。Co-IP、GST 下拉实验及免疫荧光证实,UHRF1 与 GRP78 在细胞质中直接相互作用。泛素化实验显示,UHRF1 过表达可增强 GRP78 的泛素化修饰,且特异性介导 K48 连接型泛素化(对 K63 连接型无影响),提示其通过蛋白酶体途径降解 GRP78。结构域映射实验表明,GRP78 的底物结合域(SBD,300–645 aa)是与 UHRF1 相互作用的关键区域,缺失 SBD 的 GRP78 突变体无法与 UHRF1 结合,也不能被其诱导 K48 连接泛素化。DMFold 结构预测进一步验证了 GRP78 SBD 与 UHRF1 之间的氢键相互作用,明确 UHRF1 作为 E3 泛素连接酶,通过与 GRP78 的 SBD 结合介导其泛素化降解。
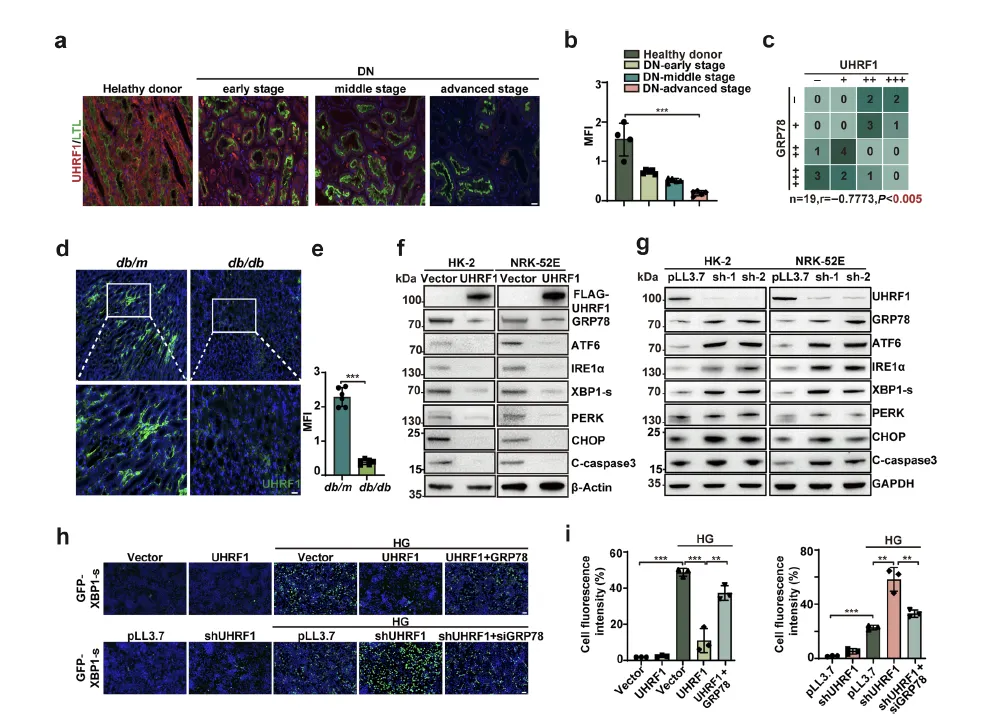
Figure 4:UHRF1 以 GRP78 依赖的方式调控肾小管上皮细胞的内质网应激和凋亡
该图验证了 UHRF1-GRP78 轴在调控 ER 应激和细胞凋亡中的功能关联性。临床样本和动物实验显示,DN 患者肾小管上皮细胞(RTECs)中 UHRF1 表达随疾病进展逐渐降低,且与 GRP78 表达呈显著负相关(r=-0.7773),db/db 小鼠肾组织中 UHRF1 水平也明显低于 db/m 对照小鼠。细胞实验中,UHRF1 过表达可降低 GRP78 水平,同时抑制 ER 应激标志物(ATF6、IRE1α、XBP1-s、PERK)和凋亡标志物(CHOP、裂解型 caspase3)的表达;反之,UHRF1 敲低则上调上述分子。XBP-1/EGFP 报告系统显示,高糖诱导的 XBP-1 剪接(ER 应激标志)可被 UHRF1 过表达抑制,且该效应能被 GRP78 过表达逆转,而 UHRF1 敲低加剧的 XBP-1 剪接可通过 GRP78 沉默恢复。蛋白复性实验证实 GRP78 具有保护变性蛋白复性的功能,而 UHRF1 可削弱该功能。关键的拯救实验显示,UHRF1 敲低诱导的 ER 应激和凋亡激活,在同时敲低 GRP78 后被完全阻断,直接证明 UHRF1 对 ER 应激和凋亡的调控依赖于 GRP78。
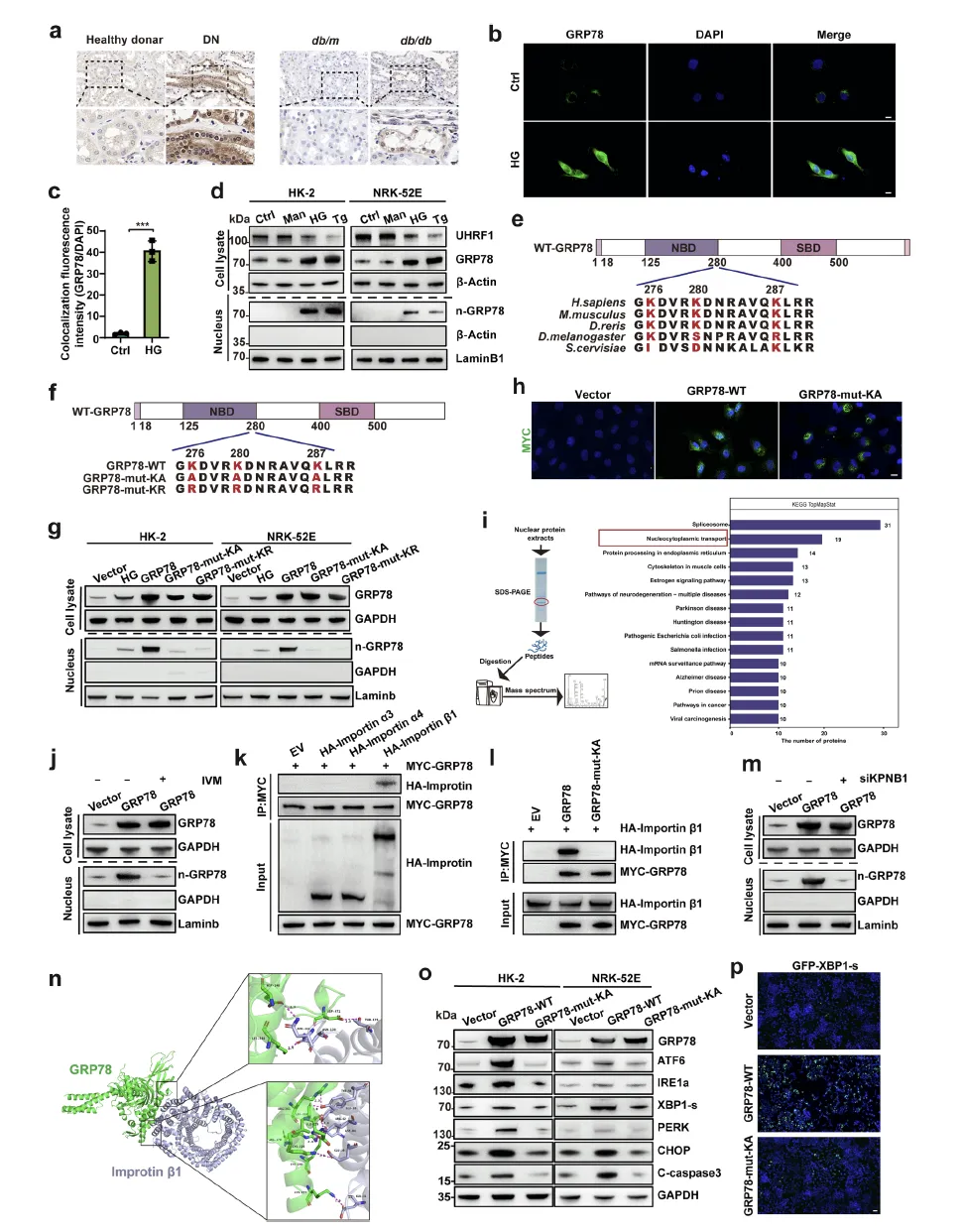
Figure 5:GRP78 积累通过 importin β1 介导的核转运加剧高糖环境下肾小管上皮细胞的内质网应激和凋亡
该图阐明了 GRP78 核转运的机制及其病理功能。免疫组化结果显示,DN 患者和 db/db 小鼠的肾小管细胞中,除细胞质外,GRP78 在细胞核内显著积累,且该现象在急性肾损伤、IgA 肾病等其他肾小管损伤疾病中也存在。细胞实验证实,高糖、转化生长因子 -β、顺铂等损伤因素可诱导 GRP78 从内质网周围向细胞核转移,核蛋白提取实验进一步验证了这一结果。UHRF1 敲低虽能增加 GRP78 核转运,但 GRP78 的 SBD 缺失突变体仍可正常核转运,且不受 UHRF1 调控,表明 UHRF1 不直接参与 GRP78 核定位。序列分析发现 GRP78 存在保守的核定位信号(NLS,276–287 aa),突变该区域的赖氨酸残基(GRP78-mut-KA/GRP78-mut-KR)可显著抑制其核转运。核提取物质谱分析结合 Co-IP 证实,GRP78 通过 NLS 与 importin β1(KPNB1)特异性结合,importin α3/α4 不参与该相互作用,且 importin β1 敲低或药物抑制(伊维菌素)能有效阻断 GRP78 核转运。功能实验显示,GRP78-WT 过表达可激活 ER 应激和凋亡通路,而核转运缺陷的 GRP78-mut-KA 则无此效应,证实 GRP78 的核定位是其诱导病理损伤的关键。
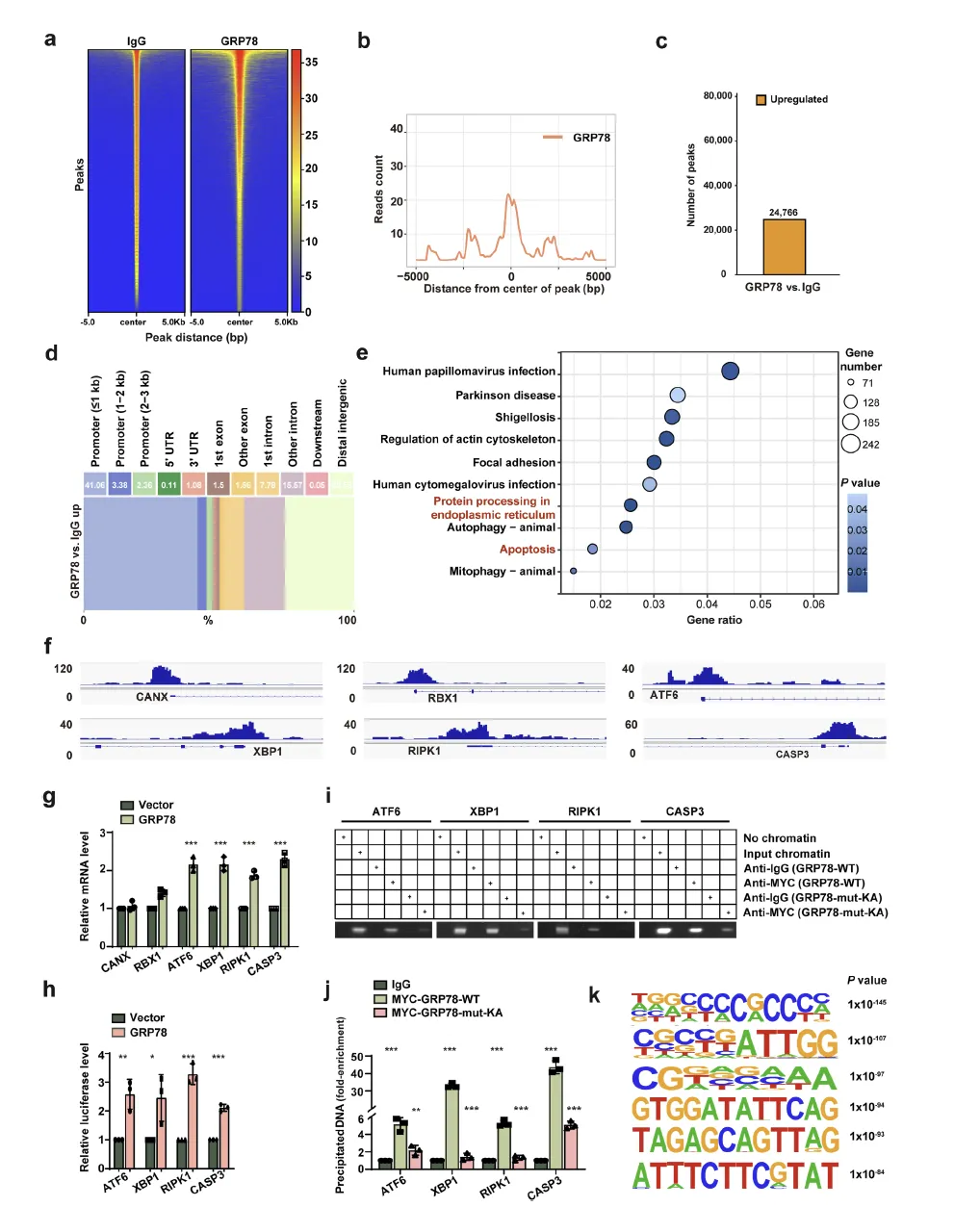
Figure 6:核定位的 GRP78 调控内质网应激和凋亡相关基因的转录程序
该图通过 CUT&Tag 技术揭示了核 GRP78 的转录调控功能。GRP78 过表达的 HK-2 细胞中,CUT&Tag 分析鉴定出 24766 个 GRP78 结合峰,其中 46.8% 定位于基因启动子区域(转录起始位点上游≤3 kb),提示 GRP78 作为转录调控因子发挥作用。KEGG 通路富集显示,GRP78 结合的启动子主要富集于 ER 蛋白加工和凋亡通路,包括 CANX、RBX1、ATF6、XBP1、RIPK1、CASP3 等关键基因。RT-qPCR 和荧光素酶报告实验证实,GRP78 过表达可显著上调这些靶基因的 mRNA 表达和启动子活性,而 importin β1 敲低能逆转该效应。ChIP 实验进一步验证,GRP78 可直接结合这些基因的启动子区域,且核转运缺陷的 GRP78-mut-KA 无法结合这些启动子。 motif 分析鉴定出 GRP78 特异性的 DNA 结合基序(如 ATTGG、CGAA 等),且其结合模式与 KLF5、KLF15 等 ER 应激 / 凋亡相关转录因子存在关联,提示 GRP78 可能通过与这些转录因子协同作用激活靶基因转录。
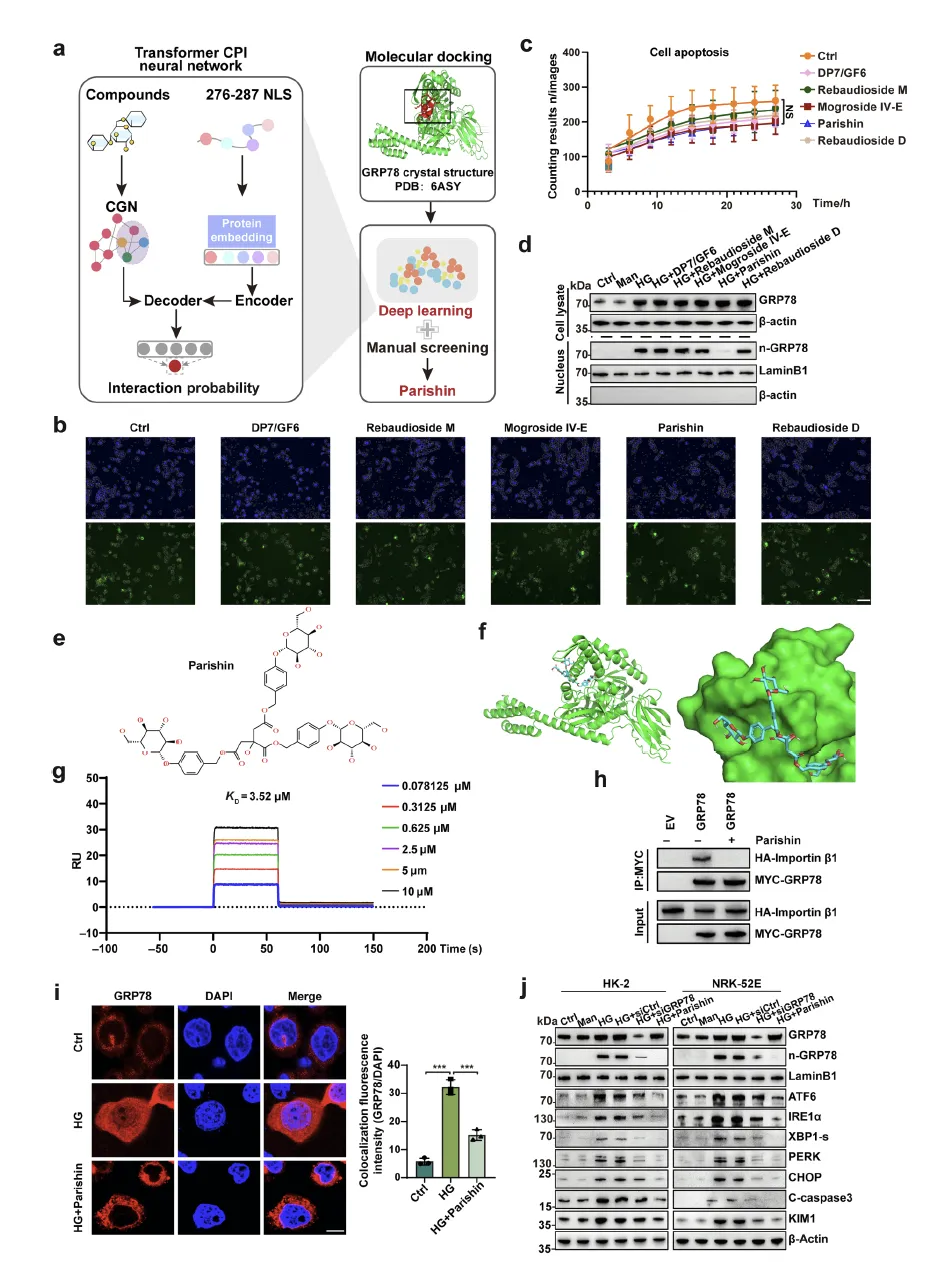
Figure 7:高通量虚拟筛选鉴定 Parishin 为 GRP78 核转运抑制剂
该图描述了 GRP78 核转运抑制剂的筛选及验证过程。采用分子对接与深度学习相结合的双平台策略,以 GRP78 的 NLS 结构域(276–287 aa)为靶点,从 210 万化合物库中筛选出 5 个候选化合物。细胞毒性实验显示,这些化合物在治疗浓度下对 HK-2 细胞活力无显著影响。Western blot 和免疫荧光验证发现,仅酚苷类化合物 Parishin 能特异性抑制高糖诱导的 GRP78 核转运,对细胞质 GRP78 水平无明显影响。SPR 分析显示 Parishin 与 GRP78 NLS 的结合亲和力为 Kd=3.52 μM,分子对接表明其通过与 NLS 域的 K280、D281、N282 残基形成氢键发挥作用。Co-IP 证实 Parishin 可减弱 GRP78 与 importin β1 的相互作用。功能实验显示,Parishin 能有效抑制高糖诱导的 ER 应激(ATF6、IRE1α 等)和凋亡(CHOP、裂解型 caspase3),且疗效优于 GRP78 敲低。蛋白复性实验证实,Parishin 处理不影响 GRP78 的细胞质分子伴侣功能,其优势在于选择性阻断 GRP78 核转运的病理作用,同时保留其生理保护功能。
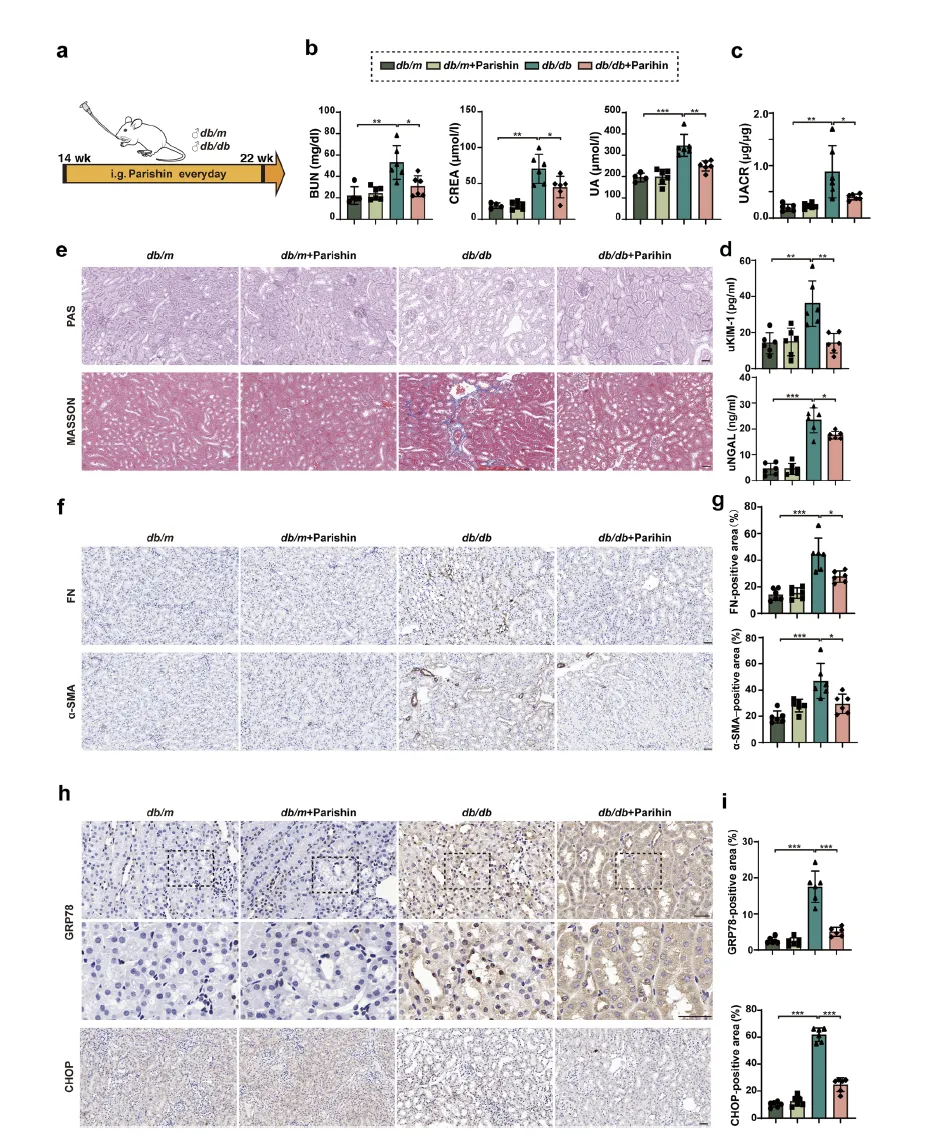
Figure 8:Parishin 通过抑制 GRP78 核转运缓解糖尿病肾病进展
该图通过动物实验验证了 Parishin 的体内治疗效果。db/db 小鼠经 Parishin 灌胃处理 8 周后,肾功能指标(血尿素氮、血清肌酐、尿酸、尿白蛋白 / 肌酐比)显著改善,尿中肾小管损伤标志物 KIM1 和 NGAL 水平降低。病理分析显示,Parishin 能有效减轻 db/db 小鼠的肾小球肥大、系膜基质扩张和肾小管萎缩,降低肾组织中纤连蛋白(FN)和 α- 平滑肌肌动蛋白(α-SMA)的表达,同时部分恢复足细胞标志物 WT-1 和突触足蛋白的表达。关键机制验证显示,Parishin 可显著抑制 db/db 小鼠肾组织中 GRP78 的核转运,同时降低 ER 应激标志物 CHOP 的表达,Western blot 和 RT-qPCR 进一步确认了相关分子的表达变化。此外,Parishin 对 db/db 小鼠的体重和糖耐量无显著影响,但可轻度改善胰岛素敏感性,表明其肾脏保护作用独立于代谢调控,主要通过抑制 GRP78 核转运、减轻 ER 应激和凋亡实现。
研究结论
本研究阐明了 DN 中 GRP78 异常调控的关键机制:高糖诱导 UHRF1 下调,破坏其对 GRP78 的启动子甲基化抑制及泛素化降解双重调控,导致 GRP78 胞质蓄积并经 importin β1 介导核转运,核内 GRP78 作为转录调控因子激活 ER 应激与凋亡通路,加剧肾小管上皮细胞损伤。筛选获得的 Parishin 可特异性阻断 GRP78 核移位,在保留其生理伴侣功能的同时改善 DN 病理损伤。该研究揭示了 UHRF1-GRP78 - 核转运轴在 DN 中的核心作用,为 DN 提供了新型治疗靶点,且 Parishin 具有良好的临床转化潜力。
局限性:未明确 GRP78 与核内转录因子的协同作用机制,缺乏临床样本中 Parishin 疗效验证。展望:后续可深入探究 GRP78 核内转录调控的分子网络,开展 Parishin 的临床前药代动力学及毒理学研究,同时探索该调控通路在其他 ER 应激相关疾病中的作用,拓展应用场景。
参考文献

国家杰青一对一答疑视频
医学省自然申请答疑,立项的关键条件是哪一些?从哪些方向可以杀出重围
临床型博士如何准备国青标书?没有预实验怎么办?专家一对一解答规划
中医药科研研究