文章于2026年1月14日发表在Journal of the American Chemical Society,南京理工大学翟傈,香港城市大学Zhang An,广东工业大学Li Meng,张山青为通讯作者。
Machine Learning-Guided Coordination Engineering of M−N−C Single-Atom Electrocatalysts for Superior Oxygen Reduction
DOI:10.1021/jacs.5c20189
摘要
单原子催化剂(SACs)的局部配位结构与其催化性能间的定量构效关系长期缺乏普适性描述符,严重制约了其理性设计。本研究通过融合密度泛函理论(DFT)高通量筛选与机器学习(ML),系统解析了312种M–N–C SAC模型(涵盖26种过渡金属与12种配位构型)的氧还原反应(ORR)活性。数据挖掘揭示了一个低维可解释描述符 Eemb/ sin(CN),(emb为单原子嵌入能,CN为配位数),该描述符同时量化金属–载体相互作用强度与几何配位环境,能够统一描述不同金属中心的ORR活性趋势。基于此设计原则,通过缺陷工程策略精准合成富含吡咯型Cu–N3配位的催化剂,其半波电位达0.886 V(vs. RHE),超越商业Pt/C(0.872 V),锌空电池峰值功率密度达191.3 mW cm−2。该工作突破了SACs“黑箱式”设计瓶颈,建立了配位工程的普适性设计范式,为下一代电催化剂的理性开发提供了可迁移的方法论框架。
研究内容
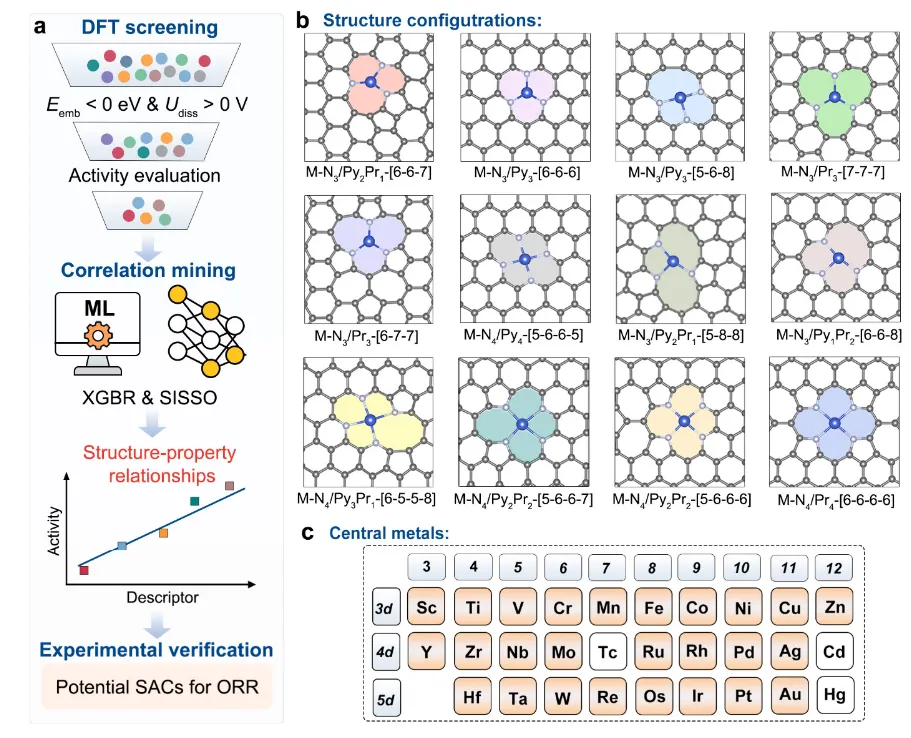
图1 过渡金属(TMs)与配位结构的理论筛选用于SACs
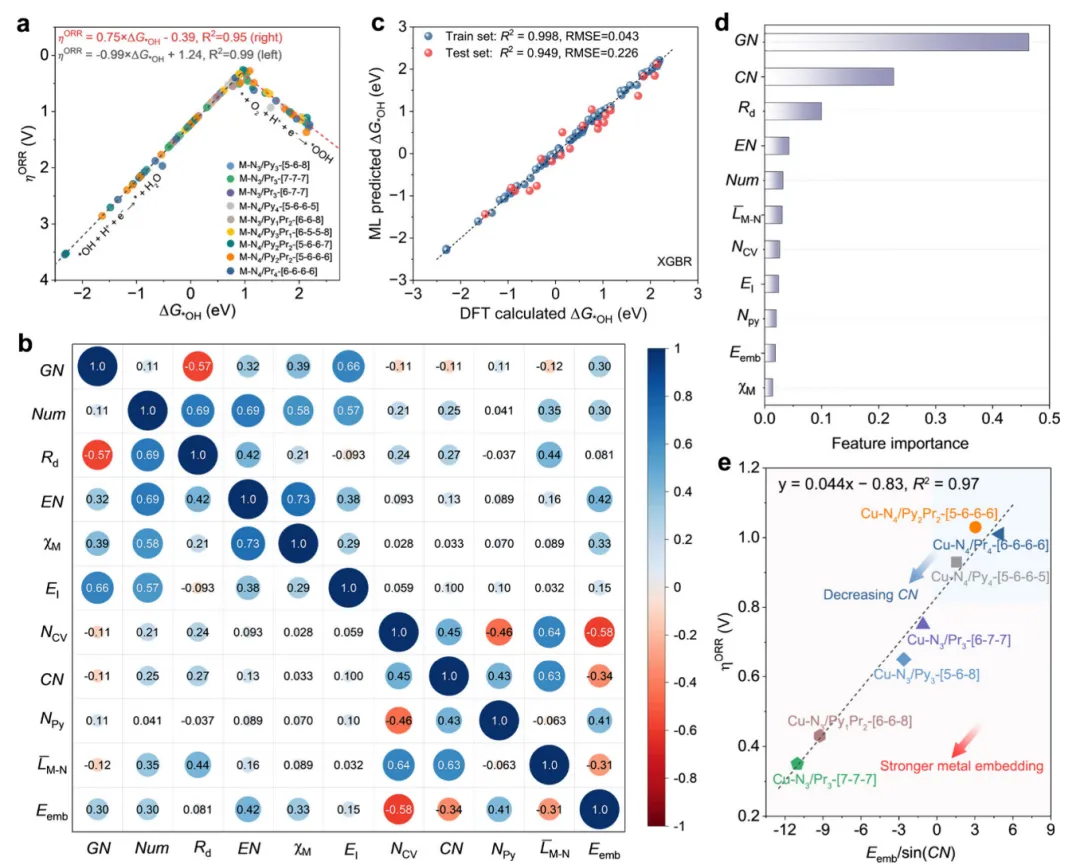
图2 DFT计算与基于机器学习的数据分析
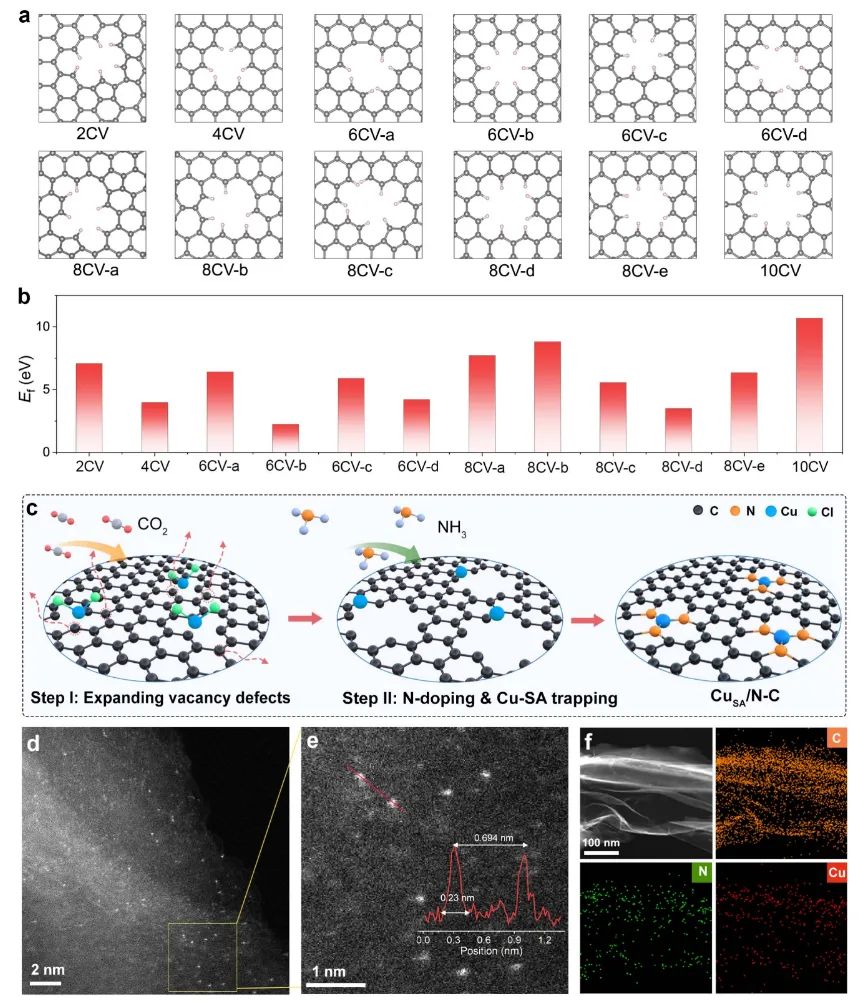
图。CuSA/N−C催化剂的设计、制备及电镜表征
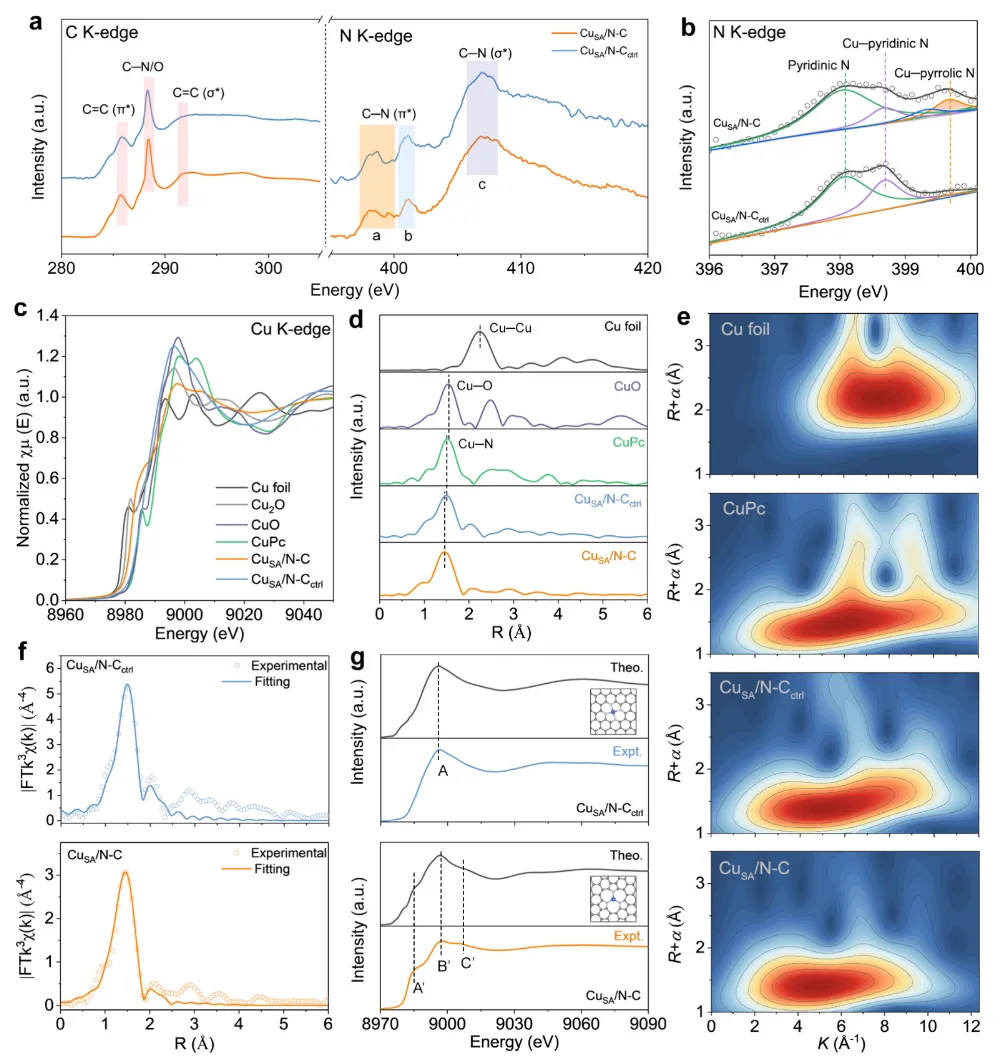
图4 XAS表征的催化剂局部化学构型
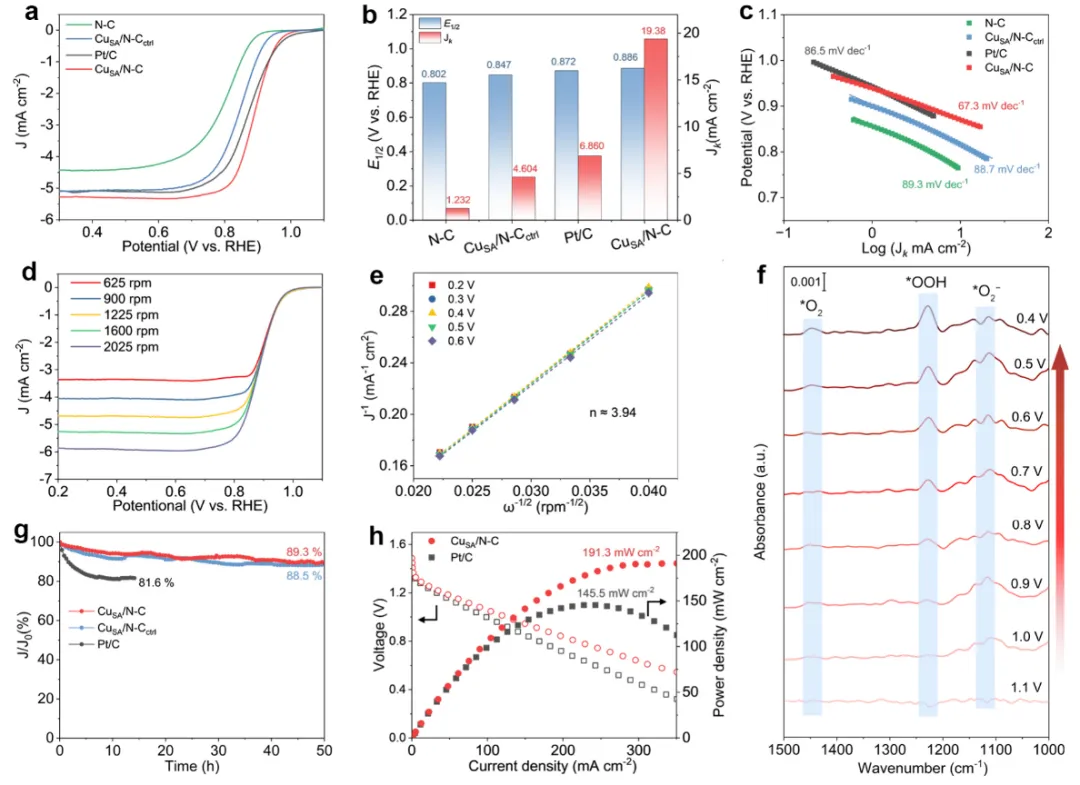
图5 催化剂的电化学及相关表征
结论与意义
本研究通过可解释机器学习破解了单原子催化剂配位结构与催化性能间的"黑箱"关系,确立了Eemb/sin(CN) 这一兼具物理意义与普适性的结构描述符,揭示了金属–载体相互作用与配位几何的协同调控机制:前过渡金属(Mn, Fe等)需通过高配位/强嵌入能削弱过强的氧吸附,而后过渡金属(Cu, Ag, Pd, Pt)则可通过降低配位数增强中间体结合,从而跨越传统"高配位更优"的认知局限。该工作不仅实现了从312个理论模型中精准提炼设计原则并指导Cu–N3催化剂的定向合成,更构建了"理论筛选–算法挖掘–缺陷工程–性能验证"的全链条理性设计范式,将单原子催化从经验试错推向可预测、可调控的新阶段。其核心价值在于:以简洁物理量统一复杂构效关系,使算法输出转化为可操作的合成策略,为高熵单原子、双原子乃至多反应体系的催化剂设计提供了可迁移的方法论框架,同时为AI驱动材料创制树立了"可解释性优先"的科学范式,对推动电催化从基础研究向能源器件实用化转化具有深远启示。
声明
本公众号专注于分享前沿领域的最新动态和科研进展。如您发现内容存在错误或涉及侵权问题,请及时通过私信与我们联系,我们会迅速核实并作出相应修正或删除处理。感谢各位读者的关注与支持!同时,我们也诚挚邀请广大科研工作者踊跃投稿,分享您的研究成果或见解。期待与您共同推动学术交流与发展!

 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
