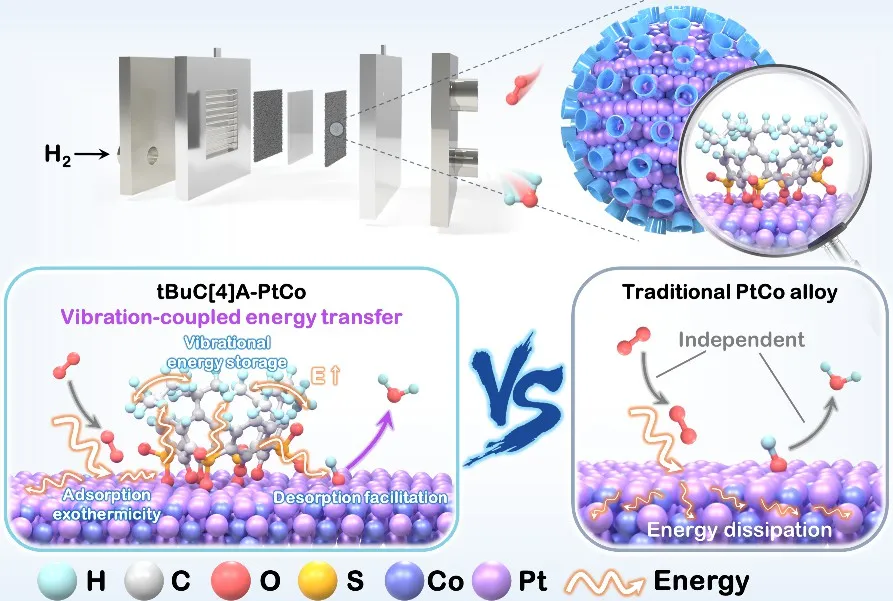
2026年01月29日,南京大学丁维平团队在Journal of the American Chemical Society期刊发表题为“Dynamic Vibration-Coupled Energy Transfer for Boosting ORR Catalysts in Fuel Cells”的研究论文,团队成员Liang Chenjia、Yao Jun、Hou Xiaoxia为论文共同第一作者,丁维平为论文通讯作者。
第一作者:Liang Chenjia、Yao Jun、Hou Xiaoxia
通讯作者:丁维平
通讯单位:南京大学
论文DOI:10.1021/jacs.5c15936
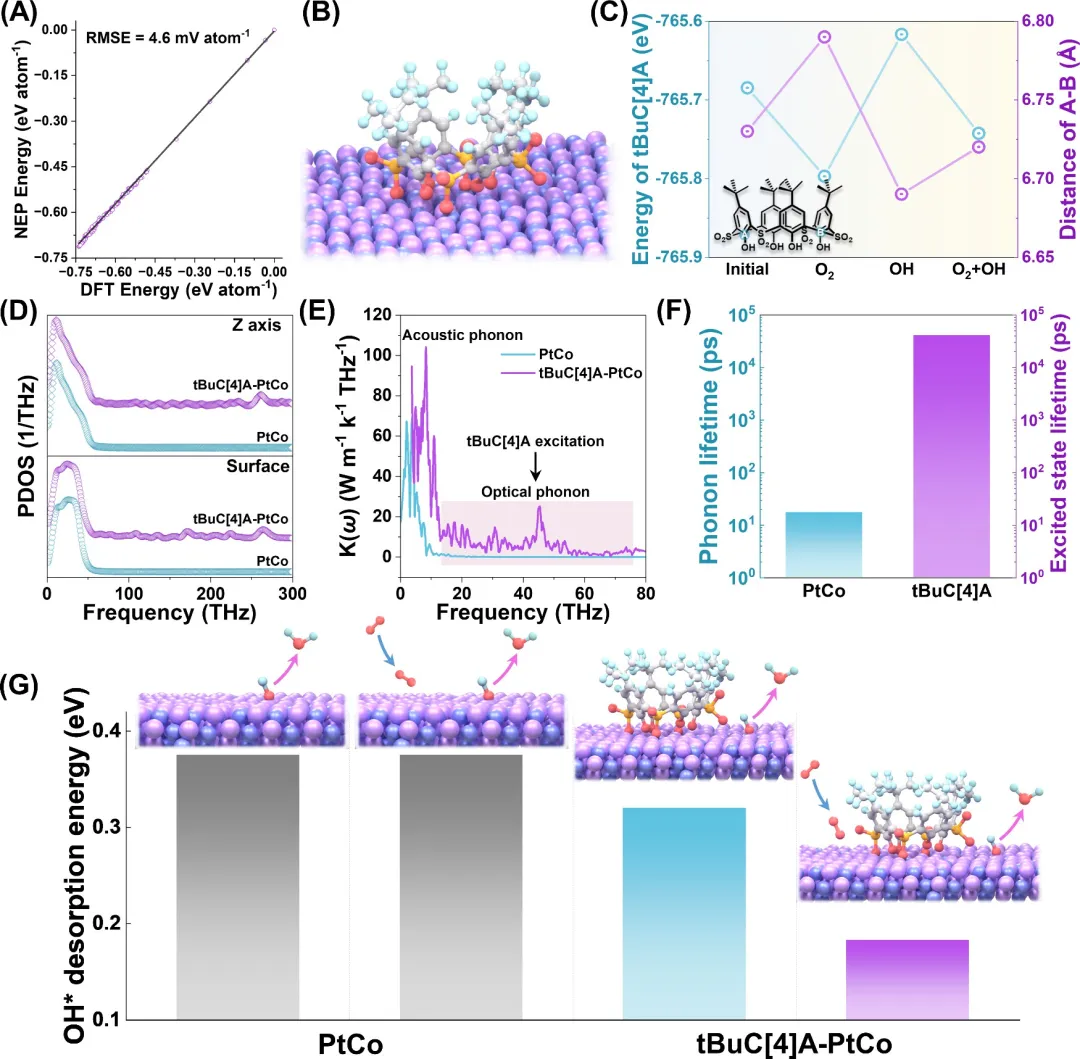
该研究展示了一种前景广阔但极具挑战性的方法,即通过动态表面能量转移,利用放热吸附来促进吸热脱附步骤,从而制备高活性催化剂。以氧还原反应(ORR)催化剂为例,研究人员通过在PtCo纳米颗粒表面锚定柔性叔丁基磺酰基杯[4]芳烃(tBuC[4]A),实现了动态振动耦合能量转移(VCET),将表面氧吸附的放热能延长保留,以补偿相邻Pt位点上诸如OH* → H₂O等吸热过程。上述过程可概括为“吸附-促进-脱附”。基于机器学习模型的理论模拟揭示,由O₂在PtCo上吸附触发并通过声子-声子散射传递的tBuC[4]A的振动激发(每分子0.126 eV),如何共振削弱OH*的键合强度,将脱附能垒降低51%(从0.375 eV降至0.183 eV),并实现了13.2%的能量利用率。这种动态能量管理显著提升了催化剂和燃料电池的性能:在0.1 mgₚₜ cm⁻²的负载量下,质量活性达到1.03 A mgₚₜ⁻¹,峰值输出功率达到2.07 W cm⁻²,超越了美国能源部的目标。原位傅里叶变换红外光谱、拉曼光谱和动力学测量等表征结果充分支持了该机理,并表明O-O键的活化与优化的OH*脱附之间存在耦合。VCET机制为超越传统策略以开发高性能多相催化剂的动态促进提供了蓝图。
质子交换膜燃料电池(PEMFC)作为一种高效的能量转换装置,由于需要使用大量贵金属催化剂来促进其大规模应用而受到限制。在过去二十年中,研究人员利用配体或应变效应开发了一系列Pt-M(M = Fe, Co, Ni等)纳米催化剂,以实现低Pt用量和高氧还原反应(ORR)活性。然而,基于其他机制的探索从未停止。
迄今为止,设计高性能催化剂的研究优先考虑了电子结构调控方法。在整个广泛认可的氧还原反应五个基本步骤中,其中两个是吸热的,特别是水脱附反应的最后一步,对催化剂活性的阻碍最大;然而,这些方法忽视了ORR过程中多步骤能量变化的时序不对称性。具体而言,放热的O₂吸附和OOH*中间体形成以及吸热的OH*物种脱附无法同时优化,这是一种热力学约束,被概括为线性标度关系:ΔG(OOH*) = ΔG(OH*) + 常数。这种关系导致传统的催化剂设计陷入火山图峰值的左侧(强吸附抑制脱附)或右侧(弱吸附阻碍O-O解离)。这一局限性也呼应了多相催化中的一个基本挑战:无法同时调控放热吸附和吸热脱附过程。因此,仅通过电子结构调制来调整催化活性的传统范式无法解决吸附和脱附能之间的固有权衡。
开发动态结构似乎是克服这些限制的关键途径。最近,该研究团队展示了一种动态结构调控策略,其中一端固定在表面的高柔性聚合物链的运动可以主动扰动乙炔半加氢过程中形成的积碳前驱体,动态且持续地活化催化剂以对抗积碳引起的活性位点堵塞,并保持长期稳定性。此外,Gulder等人报道了动态氟醇-胺超分子能够酶促催化多烯环化,Duan等人提出了引入无机钴纳米岛用于乙炔加氢的动态热管理。对于相对清晰的ORR过程,肯定可以开发一种动态催化剂调控方法,通过吸附促进脱附,利用放热步骤加速吸热步骤。
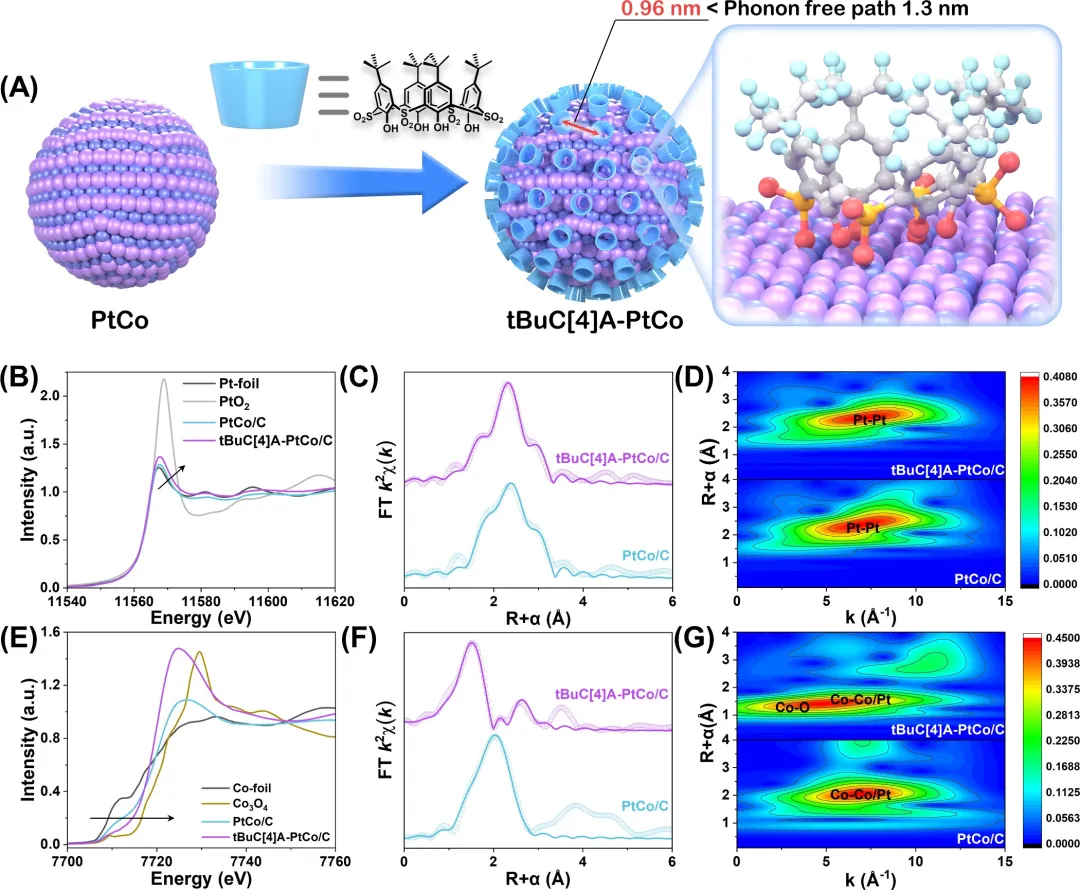
在此,该研究提出了一种通过动态振动耦合能量转移(VCET)机制实现的“吸附-促进-脱附”过程,如示意图1所示,这是从传统的电子调制向动态能量管理的范式转变。选择叔丁基磺酰基杯[4]芳烃(tBuC[4]A)作为介质分子,锚定在PtCo纳米颗粒(负载于炭黑上的PtCo纳米颗粒)上,将快速耗散的吸附能转化为加速ORR的驱动力。选择tBuC[4]A是因为它独特地结合了构象灵活性与多样的低频振动模式,以及对Co的选择性配位,从而实现对相邻Pt活性中心干扰最小的有效锚定,这是一个简单的有机小分子或刚性大环化合物很少能同时满足的协同要求。巧妙的VCET机制在于其能量管理:(1)O₂分子的初始吸附是放热的,激发表面声子-声子散射。(2)这种散射可以通过表面声子耦合传递到相邻tBuC[4]A分子的振动态,从而保留部分能量。(3)随后,一部分吸附能通过此过程在表面保留更长时间,并能够传递回表面声子,从而补偿并促进OH*脱附的吸热过程。与通过配体、应变、金属-载体相互作用静态调控电子结构,或通过微环境设计增强传质的经典策略不同,VCET机制引入了一个动态维度,管理不同基元步骤之间的能量,作为一种创新策略进一步实现高活性。
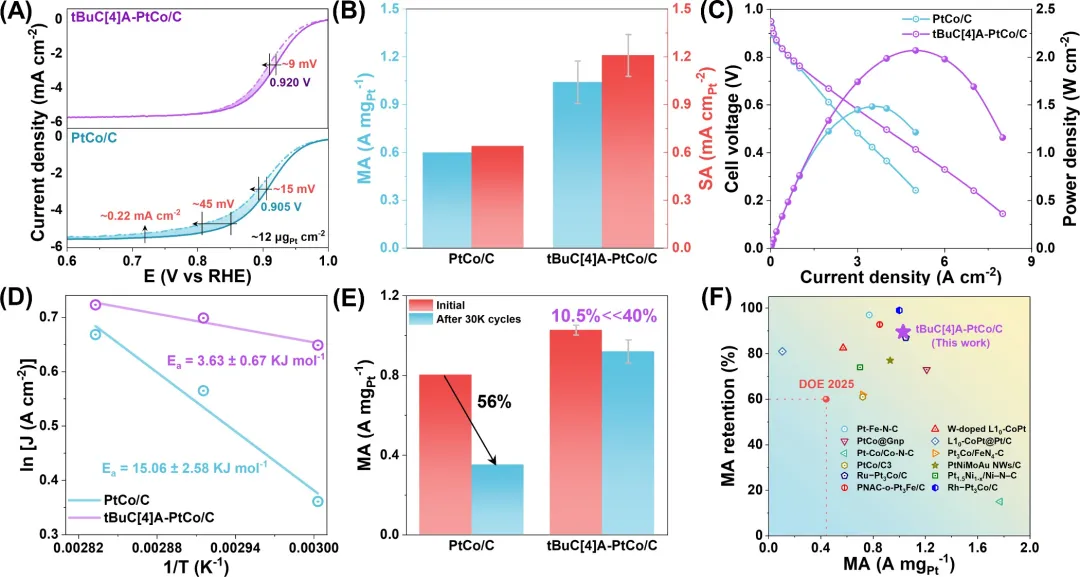
作为PEMFC的阴极,tBuC[4]A-PtCo/C的质量活性达到1.03 A mgₚₜ⁻¹(在0.9 V下),在30,000次加速降解测试(ADT)后保持率达89.5%,超过了美国能源部的目标,并且峰值功率密度达到2.07 W cm⁻²,是商业PtCo催化剂的1.4倍。tBuC[4]A-PtCo/C体系卓越的耐久性源于双重稳定效应:催化剂本身和整个膜电极组件(MEA)。基于机器学习模型的DFT计算从理论上充分描述了由tBuC[4]A分子振动之间的能量转移驱动的O₂吸附和OH*脱附的耦合。研究发现,OH* + H* + e⁻ → H₂O的吉布斯自由能垒从PtCo的0.375 eV降低到tBuC[4]A-PtCo的0.183 eV,电化学原位光谱检测ORR过程中表面物种的变化以及电极动力学测量也支持这一结果。通过声子态密度、速度自相关函数和谱热导率[K(ω)]的协同分析,全面解析了表面能量转移过程。这种能量转移策略为设计能够实时能量再分配的催化系统建立了蓝图,是向高功率、耐用PEMFC迈出的关键一步。
示意图1. 振动耦合能量转移(VCET)机理示意图。
图1. VCET机制的理论预测。(A) 训练数据集的RMSE图。(B) 在Pt₁Co₁(111)结构上优化的tBuC[4]A。(C) Pt₁Co₁(111)上tBuC[4]A的单点能和环距随不同物种吸附的变化。(D) Pt₁Co₁(111)与tBuC[4]A-Pt₁Co₁(111)之间声子态密度的比较。(E) PtCo和tBuC[4]A-Pt₁Co₁(111)的谱热导率[K(w)]。(F) tBuC[4]A的激发态寿命与Pt₁Co₁(111)声子寿命的比较。(G) 关键的OH*脱附能比较。
图2. tBuC[4]A-PtCo/C的制备与结构表征。(A) 通过tBuC[4]A键合对PtCo进行特异性修饰的示意图。(B) 归一化的Pt L₃边XANES谱。(C) Pt L₃边的EXAFS谱。(D) Pt L₃边EXAFS谱的小波变换。(E) 归一化的Co K边XANES谱。(F) Co K边的EXAFS谱。(G) Co K边EXAFS谱的小波变换。
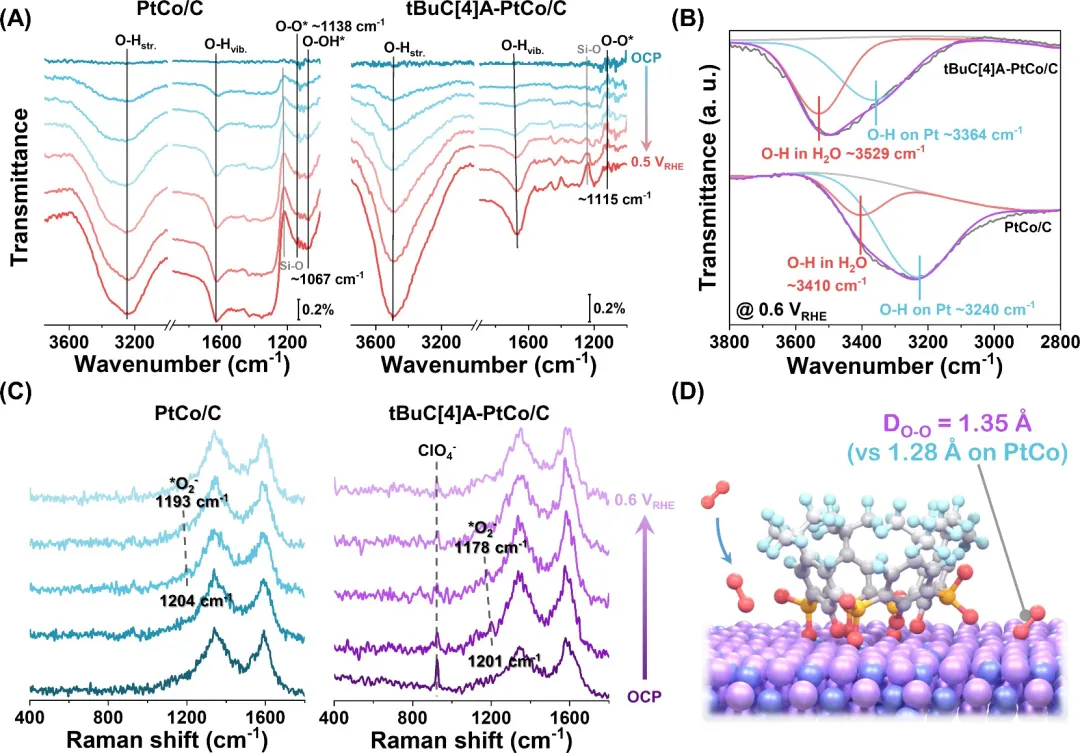
图3. ORR过程中表面物种演化的探索。(A) 室温下在O₂饱和的0.1 M HClO₄溶液中的电化学原位FTIR谱。(B) 在0.6 VRHE下O-H伸缩振动区域的峰拟合。(C) 室温下在O₂饱和的0.1 M HClO₄溶液中的电化学原位拉曼谱。(D) O₂吸附促进O₂活化的示意图。
图4. tBuC[4]A-PtCo/C的ORR性能。(A) 在O₂饱和的0.1 M HClO₄中,30,000次循环ADT前后测得的ORR极化曲线。(B) 在0.9 V(vs RHE)下,PtCo与tBuC[4]A-PtCo/C的质量活性(MA)和比表面积活性(SA)比较。(C) 分别以PtCo/C和tBuC[4]A-PtCo/C为阴极催化剂的PEMFC的极化曲线和功率密度曲线(铂负载量 ≈ 0.1 mgₚₜ cm⁻²)。(D) PtCo/C和tBuC[4]A-PtCo/C在0.8 V下的活化能。(E) 在0.9 ViR‑free条件下,PtCo/C与tBuC[4]A-PtCo/C在30,000次循环ADT前后的质量活性比较。(F) 初始质量活性与30,000次循环ADT后活性保持率与近期报道的典型阴极电催化剂的比较。
总之,该研究通过振动耦合能量转移机制展示了一种动态活化催化剂,作为打破ORR标度关系的范式。通过构建tBuC[4]A-PtCo界面,将O₂吸附能(-0.956 eV)转化为tBuC[4]A分子振动,约13.2%的吸附能参与到动态活化体系中,将附近位点OH*的脱附能垒降低至0.183 eV。通过声子态密度、速度自相关函数和谱热导率详细描述了能量转移过程。在实际PEMFC中,该催化剂在0.1 mgₚₜ cm⁻²负载下实现了1.02 A mgₚₜ⁻¹的质量活性(达到美国能源部目标的2.3倍)和2.07 W cm⁻²的峰值输出功率。原位光谱表征验证了由于振动耦合能量转移,O₂在tBuC[4]A-PtCo/C上的吸附加速了OH*脱附。此外,tBuC[4]A修饰也增强了PtCo基催化剂的稳定性,在30,000次循环ADT后保持了89.5%的初始活性。VCET机制通过尽可能利用吸附热来促进缓慢且吸热的OH*脱附步骤,从而促进了ORR的催化中心。除了ORR催化剂,通过能量共振动态活化催化剂的原理,也将在受能量时序不对称性困扰的多步反应催化剂中展现出变革潜力。