四校联合AM!南京大学「国家优青」金钟&合肥工业大学许海峰&巢湖大学王雯最新AM:调控α-MoC₁₋ₓ负载钌团簇与单原子实现高效水分解!
Precision-Engineered Electronic Modulation of Ruthenium Clusters and Single Atoms on Vacancy-Rich α-MoC₁₋ₓ Enables Efficient Electrocatalytic Water Splitting
在富空位α-MoC₁₋ₓ载体上对钌团簇及单原子进行精准电子调控,可实现高效电催化水分解https://doi.org/10.1002/adma.202519840在保持高效催化活性的同时,最大化活性金属的利用率对于电催化碱性析氢反应至关重要。本研究提出一种简易热解策略,将钌(Ru)团簇与邻近的Ru单原子锚定于α-MoC₁₋ₓ包覆的碳纳米球表面(命名为RuCS/SA/α-MoC₁₋ₓ/C)。理论计算结合原位表征揭示,通过电子桥接机制,Ru单原子向缺陷型α-MoC₁₋ₓ捐赠电子,后者再将电子转移至Ru团簇,实现对不同类型Ru位点电子结构的协同调控。因此,Ru单原子与α-MoC₁₋ₓ的双重激发作用削弱了Ru团簇与H*的结合强度,加速了H₂的脱附。所制备的3%-RuCS/SA/α-MoC₁₋ₓ/C样品在10 mA cm⁻²电流密度下仅需9 mV过电位,质量活性达20.38 A mg⁻¹Ru(-100 mV),25 mV过电位下的转化频率为1.71 H₂ s⁻¹,均优于20% Pt/C催化剂。此外,采用该材料作为阴极电催化剂的阴离子交换膜水电解槽及锌-水电池均展现出优异性能。氢能作为最具潜力替代化石燃料的绿色能源之一,对减少空气污染和缓解气候变化具有重要意义。目前,电催化析氢反应(HER)被视为最清洁的制氢技术,其可利用太阳能、风能和潮汐能等可再生能源产生的绿色电力可持续制取氢气(H₂)。作为经济高效生产高纯度氢气的关键工业方法,碱性HER已成为科学研究的热点。然而,碱性HER的制氢效率低于酸性HER,主要源于其特有的Volmer步骤(H₂O + e⁻ → Had + OH⁻),该步骤被认为是碱性HER的速率决定步骤。尽管铂(Pt)基贵金属催化剂在碱性HER领域取得显著进展,但其高成本仍是制约实际应用的关键瓶颈。因此,开发高效廉价的碱性HER催化剂至关重要。钌(Ru)的成本仅为铂的约30%,且与Pt具有相近的水结合能,被视为HER中Pt的理想替代材料。迄今为止,虽然Ru基电催化剂在酸性HER中表现出色,但在碱性HER中的催化活性仍受限于缓慢的水解离过程,导致性能欠佳。其根本挑战在于质子供应不足,源于Volmer步骤中H₂O解离迟缓及生成的OH*吸附过强,二者协同作用引发催化剂中毒。因此,设计高效Ru基电催化剂以实现碱性HER性能媲美酸性HER,仍是亟待解决的关键难题。
近期,He等研究者发现,嵌入镍空位的Ru单原子可引发Ru位点局部结构极化,降低水解离能垒并优化氢吸附自由能,使碱性HER在10 mA cm⁻²电流密度下过电位降至54 mV。此外,Hu等研究者揭示了Ru团簇的电子结构具有尺寸依赖性:1 nm Ru团簇较单原子展现出更强的H₂O解离能力及d带中心上移特性,而3 nm Ru团簇因氢吸附-脱附动力学优化而具有更高的HER活性。Zhang等进一步提出,LaRuSi₃表面电化学触发的Ru团簇再生可诱导电荷去局域化,促进Ru活性位点的水吸附-解离,同时增强导电性和电化学表面积。这些研究共同揭示了钌基材料在碱性HER中独特的尺寸效应及界面调控机制。鉴于此,我们推测Ru团簇与单原子协同作用可能更有利于水解离。然而,OH*中毒问题及单原子易团聚成大团簇的倾向仍是亟待克服的挑战。
大量研究表明,适宜的载体不仅能锚定金属原子/团簇以抑制团聚,还可通过强金属-载体相互作用(SMSI)优化负载金属的电子结构[,从而调控OH*吸附能垒以减轻中毒效应。金属碳化物具有优异的稳定性、导电性和耐腐蚀性。此外,碳的电负性低于O、S、Se和P等元素,有助于优化Ru的电子结构,使更多d电子参与HER。值得注意的是,富含空位的α-MoC₁₋ₓ具有本征HER活性,展现出巨大潜力。理论分析表明,α-MoC₁₋ₓ可显著加速Volmer步骤动力学,从而提升HER速率。然而,受限于α-MoC₁₋ₓ的合成挑战,利用其作为载体锚定Ru单原子和团簇的探索性研究极为匮乏,导致二者协同机制尚未阐明。
本研究提出一种简易热解策略,成功将Ru团簇与邻近单原子锚定于α-MoC₁₋ₓ载体表面。得益于碳框架的空间限域与保护作用,尺寸约5 nm的α-MoC₁₋ₓ纳米晶被紧密包裹于碳基质中。富含空位的α-MoC₁₋ₓ作为独特载体,不仅稳定了Ru单原子和团簇(命名为RuCS/SA/α-MoC₁₋ₓ/C),还通过丰富的缺陷位点直接参与电荷再分配。高角环形暗场扫描透射电子显微镜(HAADF-STEM)结果证实,Ru团簇与单原子成功锚定于α-MoC₁₋ₓ表面。密度泛函理论(DFT)计算首次预测了电子桥接机制:Ru单原子向缺陷型α-MoC₁₋ₓ捐赠电子,后者再将电子转移至Ru团簇,从而实现对不同类型Ru位点电子结构的协同调控。X射线光电子能谱(XPS)和X射线吸收精细结构谱(XAFS)分析进一步验证了DFT预测的单原子与团簇间相反的电荷转移方向。通过系统调控钌负载量,本研究确定3%为单原子与团簇平衡共存的最佳组成比例。所制备的3%-RuCS/SA/α-MoC₁₋ₓ/C在碱性溶液中表现出优异性能:10 mA cm⁻²电流密度下过电位仅9 mV,质量活性达20.38 A mg⁻¹ Ru,25 mV过电位下转化频率(TOF)为1.71 s⁻¹,均显著优于20% Pt/C催化剂。以3%-RuCS/SA/α-MoC₁₋ₓ/C为阴极催化剂的阴离子交换膜水电解槽(AEMWE)在1.0 A cm⁻²电流密度下稳定运行550 h。此外,基于3%-RuCS/SA/α-MoC₁₋ₓ/C构建的1 m KOH体系锌-水电池实现持续发电,最大功率密度达22.50 mW cm⁻²,并在10 mA cm⁻²电流密度下可持续放电20 h,这得益于3%-RuCS/SA/α-MoC₁₋ₓ/C催化剂卓越的碱性HER性能。本研究不仅通过反向捕获策略在原子层面有效提升了催化剂活性,还为构建高效AEMWE和锌-水电池开辟了新途径。
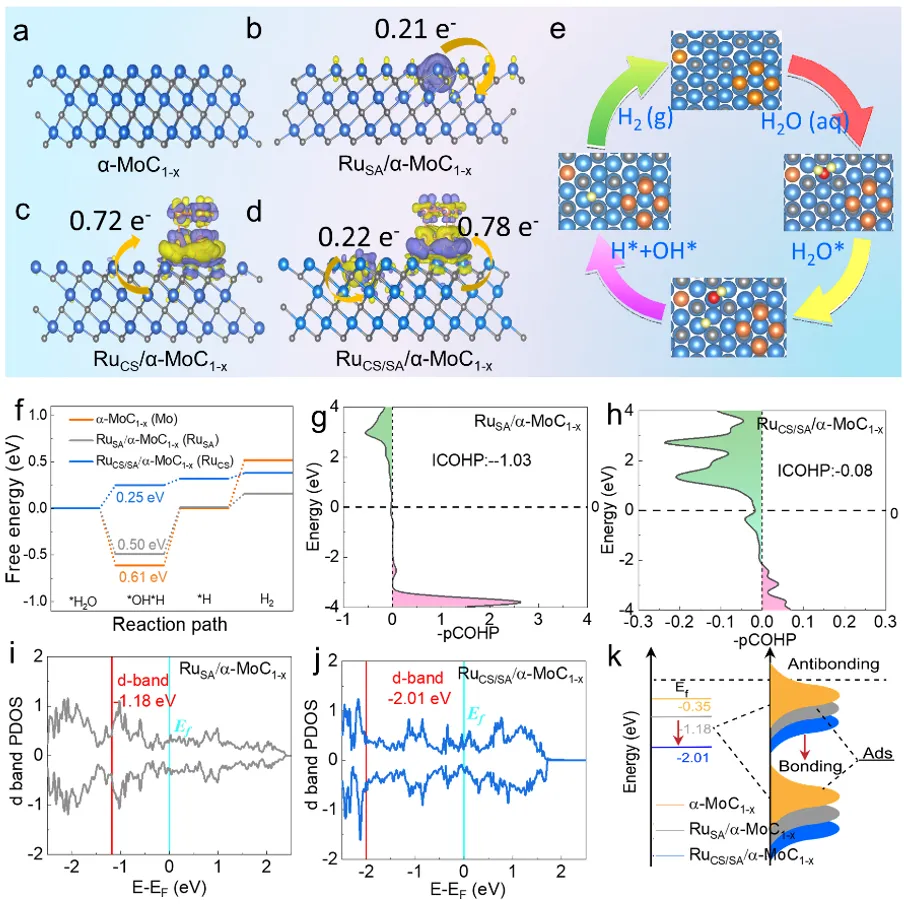
图1a-d) α-MoC₁₋ₓ、RuSA/α-MoC₁₋ₓ、RuCS/α-MoC₁₋ₓ和RuCS/SA/α-MoC₁₋ₓ的差分电荷密度(等值面单位为0.007 eÅ⁻³,浅蓝色、灰色和橙色球分别代表Mo、C和Ru)。e) 主要反应步骤的中间产物。f) α-MoC₁₋ₓ、RuSA/α-MoC₁₋ₓ和RuCS/SA/α-MoC₁₋ₓ/C模型上完整HER反应的反应能剖面。g和h) RuSA/α-MoC₁₋ₓ和RuCS/SA/α-MoC₁₋ₓ的积分晶体轨道哈密顿布居(ICOHP)值。i,j) RuSA/α-MoC₁₋ₓ和RuCS/SA/α-MoC₁₋ₓ/C的d带投影态密度(PDOS)。k) α-MoC₁₋ₓ、RuSA/α-MoC₁₋ₓ和RuCS/SA/α-MoC₁₋ₓ/C的d带中心。
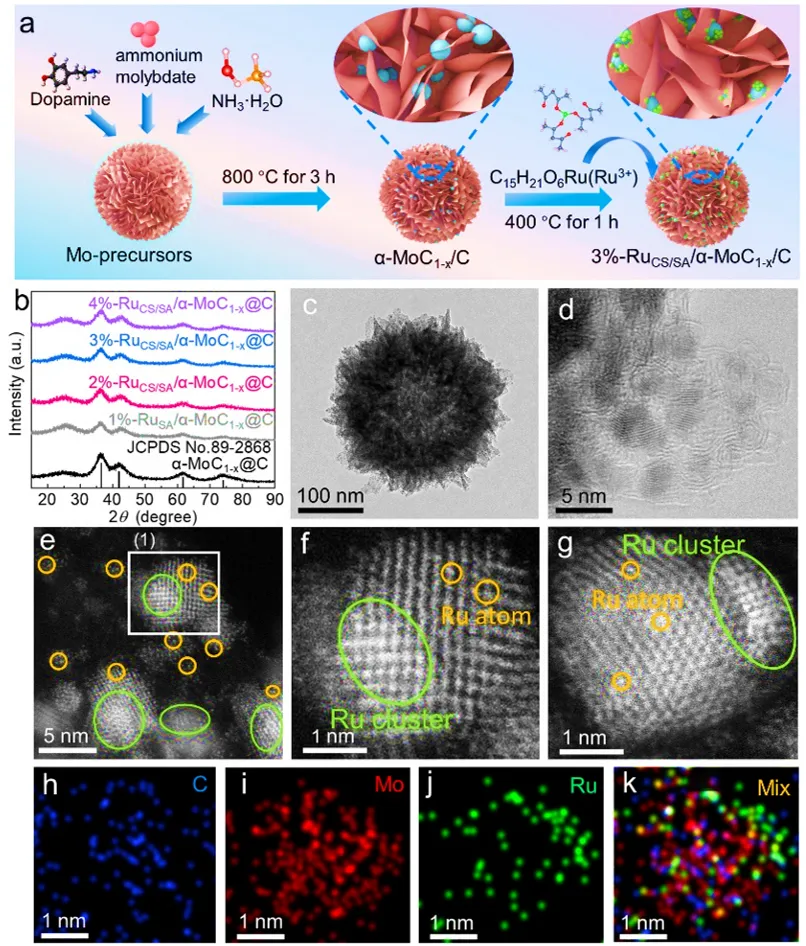
图2a) 3%-RuCS/SA/α-MoC₁₋ₓ/C的合成示意图(粉色、蓝色和绿色球分别对应C、Mo和Ru元素)。b) x%-RuCS/SA/α-MoC₁₋ₓ/C的X射线衍射(XRD)图。c,d) 不同尺度下α-MoC₁₋ₓ/C的透射电子显微镜(TEM)和高角环形暗场扫描透射电子显微镜(HAADF-STEM)图像。e,f) 不同尺度下3%-RuCS/SA/α-MoC₁₋ₓ/C的HAADF-STEM图像。g) 选区电子衍射图。h-j) 3%-RuCS/SA/α-MoC₁₋ₓ/C的元素映射图像。k) C、Ru和Mo的重叠元素映射图像。
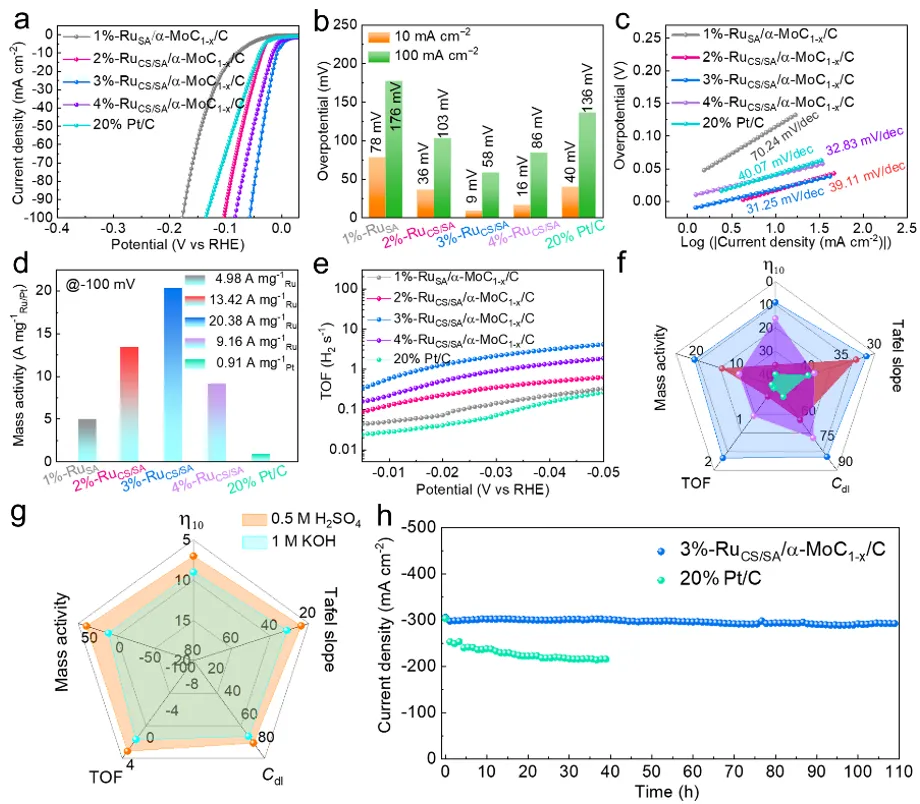
图3a) 3%-RuCS/SA/α-MoC₁₋ₓ/C与20 wt% Pt/C及其他不同Ru负载量样品的极化曲线对比。b) 电流密度为10 mA cm⁻²和100 mA cm⁻²时的过电位。c) 由对应HER极化曲线导出的塔菲尔(Tafel)图。d) 相对于参比样品在-100 mV(vs RHE)下的质量活性。e) 相对于参比样品(不同Ru负载量)的转化频率(TOF)曲线。f) x%-RuCS/SA/α-MoC₁₋ₓ/C(x%代表2%、3%或4%的Ru负载比)与商用Pt/C电催化剂的HER性能指标对比。g) 3%-RuCS/SA/α-MoC₁₋ₓ/C在碱性电解质和酸性电解质(0.5 M H₂SO₄)下的HER性能指标对比。h) 3%-RuCS/SA/α-MoC₁₋ₓ/C与20% Pt/C在300 mA cm⁻²下的计时安培法测试(I-T)。
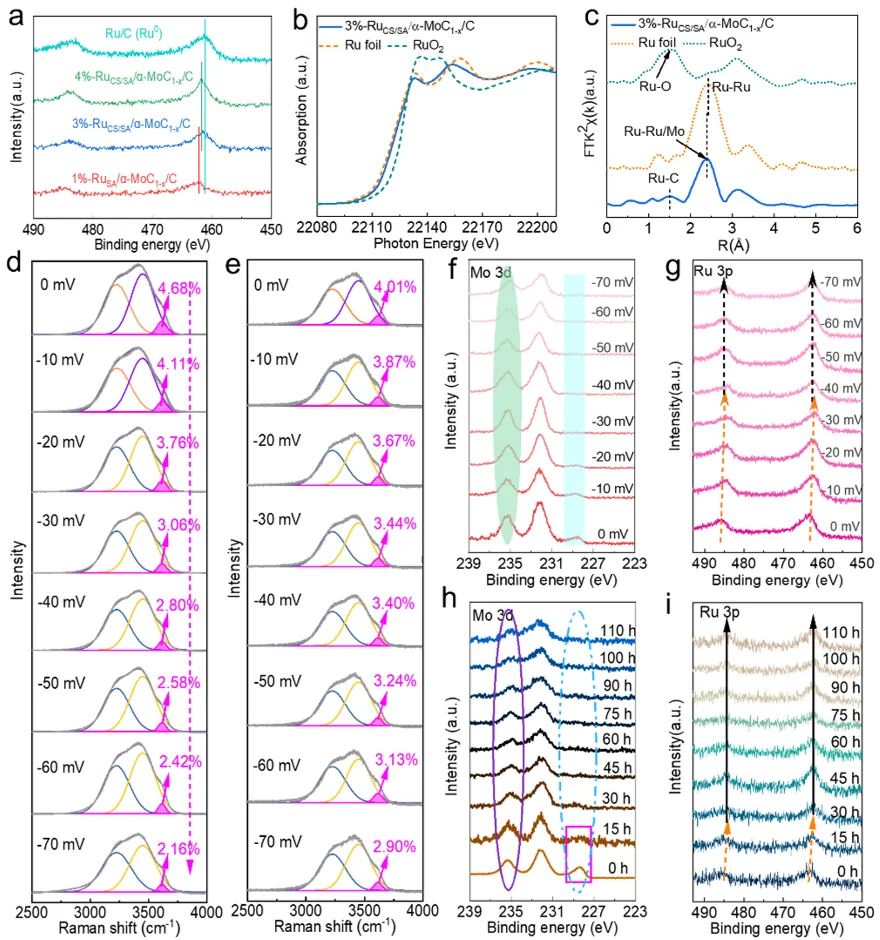
图4a) Ru 3p轨道的X射线光电子能谱(XPS)图。b) 3%-RuCS/SA/α-MoC₁₋ₓ/C、Ru箔和RuO₂的Ru K边X射线吸收近边结构(XANES)谱。c) 对应的Ru K边扩展X射线吸收精细结构(EXAFS)谱的傅里叶变换结果。d) 1%-RuSA/α-MoC₁₋ₓ/C界面水信号的原位拉曼光谱。e) 3%-RuCS/SA/α-MoC₁₋ₓ/C界面水信号的原位拉曼光谱。f,g) HER过程中3%-RuCS/SA/α-MoC₁₋ₓ/C的准原位Mo 3d和Ru 3p XPS谱分析。h,i) HER不同阶段的Mo 3d和Ru 3p XPS谱。
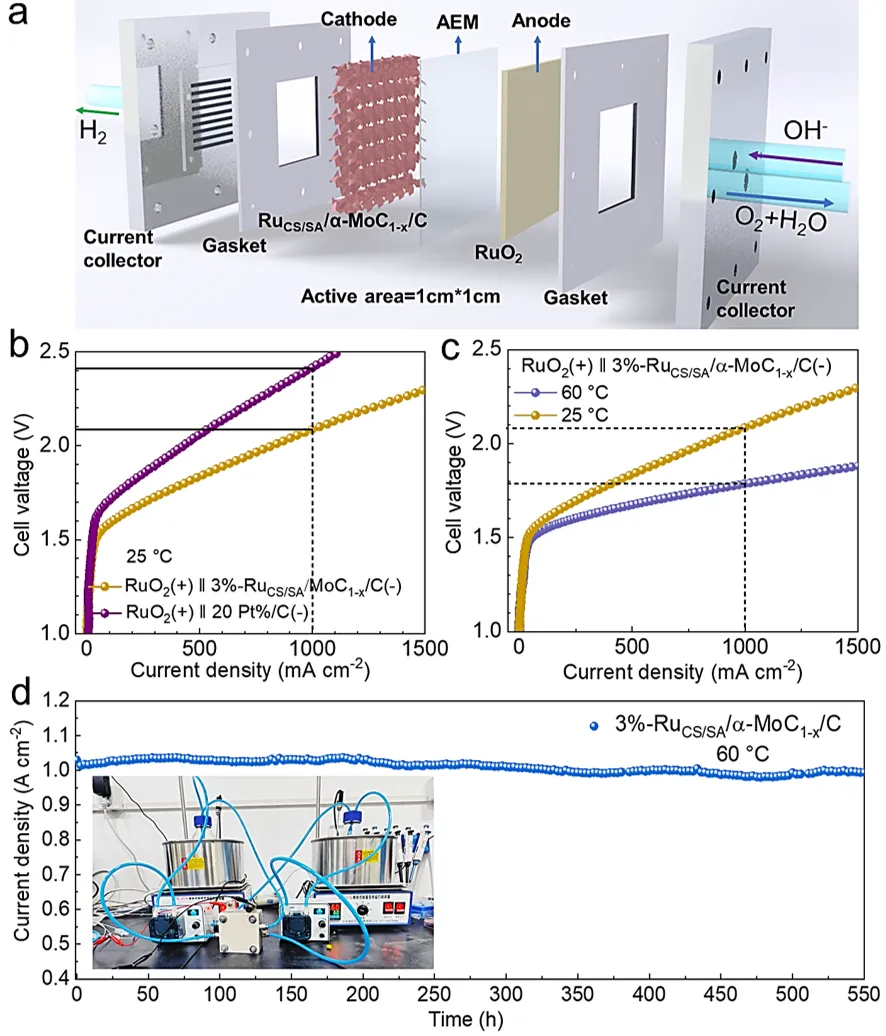
图5a) 阴离子交换膜水电解(AEMWE)装置示意图及碱性介质中AEMWE制氢示意图。b) 以3%-RuCS/SA/α-MoC₁₋ₓ/C和商用20% Pt/C为阴极催化剂的AEMWE在25℃下的极化曲线。c) 以3%-RuCS/SA/α-MoC₁₋ₓ/C为阴极催化剂的AEMWE在60℃和25℃下的极化曲线。d) 基于阴离子交换膜(AEM)的电解槽中3%-RuCS/SA/α-MoC₁₋ₓ/C在60℃下的耐久性测试。
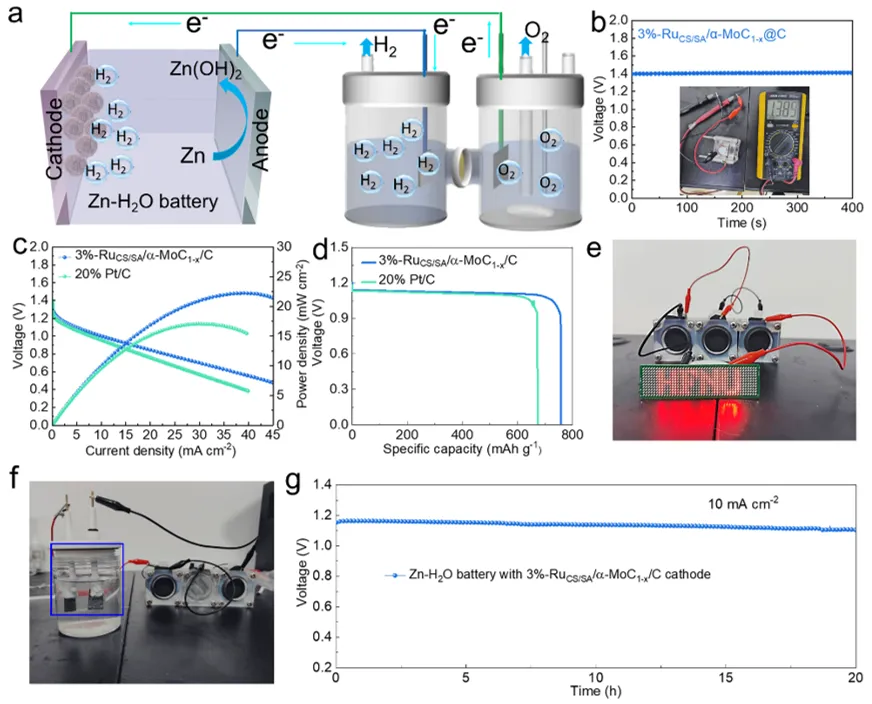
图6a) 双锌-水电池耦合配置驱动电催化水分解的示意图。b) 配备3%-RuCS/SA/α-MoC₁₋ₓ/C的锌-水电池的开路电位(OCP)。c) 配备3%-RuCS/SA/α-MoC₁₋ₓ/C的锌-水电池的线性扫描伏安曲线(LSV,左纵轴)及功率密度(右纵轴)。d) 3%-RuCS/SA/α-MoC₁₋ₓ/C与20% Pt/C的电压-比容量关系图。e) 由三个串联的装配式锌-水电池(采用3%-RuCS/SA/α-MoC₁₋ₓ/C阴极)点亮的十几个LED灯的照片。f) 由三个串联的锌-水燃料电池供电的水电解槽中阴极析氢反应(HER)和阳极析氧反应(OER)的数码照片。g) 采用3%-RuCS/SA/α-MoC₁₋ₓ/C的锌-水电池的长期耐久性测试。
综上所述,本研究通过简易热解策略成功合成出负载于α-MoC₁₋ₓ基底上、带有相邻活性Ru位点的Ru团簇,该材料作为高效碱性析氢反应(HER)电催化剂表现出色。得益于精巧的纳米反应器结构以及Ru单原子/团簇与α-MoC₁₋ₓ间独特的局部电荷再分配效应,所制备的3%-RuCS/SA/α-MoC₁₋ₓ/C催化剂展现出优异的碱性HER性能:在10 mA cm⁻²电流密度下仅需9 mV过电位,塔菲尔斜率低至31.25 mV dec⁻¹;在-25 mV过电位下具有卓越的质量活性(20.38 A mg⁻¹Ru)和转化频率(TOF,1.71 s⁻¹),同时具备长期稳定性(在300 mA cm⁻²电流密度下稳定运行110小时)。值得注意的是,采用RuO₂(阳极)和3%-RuCS/SA/α-MoC₁₋ₓ/C(阴极)的阴离子交换膜电解槽在1000 mA cm⁻²工业级电流密度下连续运行550小时,性能显著优于商用20% Pt/C催化剂。同时,自供能锌-水电池体系达到22.50 mW cm⁻²的功率密度,并在20小时内保持10 mA cm⁻²的稳定放电性能。本研究不仅为高效钌基催化剂的合成提供了宝贵策略,更从构效关系角度深化了对碱性HER催化机理的认识。








 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
