【JACS】南京大学潘惠杰/厦门大学王斌举:人工光酶催化不对称[2π+2σ]环加成反应构建手性双环[2.1.1]己烷
- 2026-06-13 04:43:52

导语
双环[2.1.1]己烷(BCHs)是具有三维结构的邻位/间位二取代苯的生物电子等排体,凭借其独特的刚性骨架结构、优异的代谢稳定性和可引入多个手性中心的特性,在药物化学中展现出巨大应用潜力。然而,这类手性骨架的高效不对称合成一直是有机合成领域的难点,尽管已有Lewis酸、过渡金属及有机光催化等方法但仍然存在着底物适用范围窄、立体选择性有限、背景反应严重等问题。
近日,南京大学潘惠杰课题组与厦门大学王斌举课题组合作在Journal of the American Chemical Society上发表研究,开发了一种基于NAD⁺类似物的人工光酶催化体系,首次实现了喹啉-2(1H)-酮与单取代双环丁烷(BCBs)的高对映选择性分子间[2π+2σ]环加成反应,核心思路是基于本课题组前期开发了一种光活性辅因子BpAD(二苯甲酮腺嘌呤二核苷酸),该NAD⁺类似物通过将烟酰胺基团替换为光敏剂实现功能改造。BpAD的引入可有效地将多种NAD⁺结合蛋白重塑为人工光酶,这些人工光酶可通过能量转移机制实现[2+2]环加成反应(Nature Catalysis 2025, 8,822)。通过对光活性辅因子BpAD进行结构修饰,以调控其光物理性质,引入给电子基团(如甲氧基)设计出MeO-BpAD,使其最大吸收红移至400 nm区域,有望在抑制底物背景反应的同时维持与各种NAD⁺依赖型蛋白质的特异性结合。将这种“可调”的光敏辅因子与工程化的蛋白质骨架结合,可重塑出一系列人工光酶,利用蛋白活性口袋的精准调控能力,实现高对映选择性的BCHs合成(图1)。
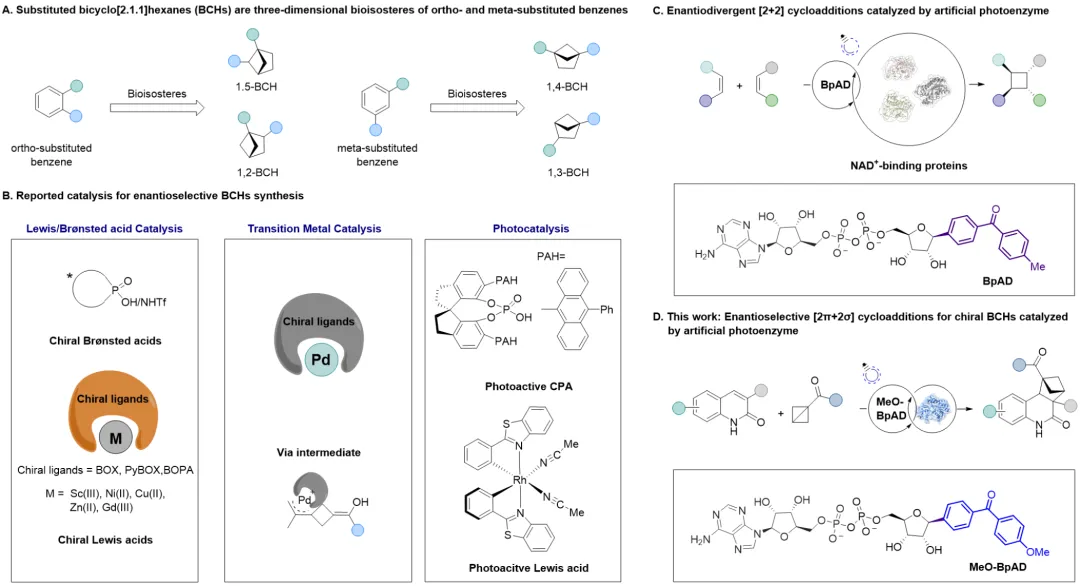
图1. 双环[2.1.1]己烷(BCHs)的对映选择性合成(图片来源:J. Am. Chem. Soc.)
前沿科研成果
首先研究者以喹啉-2(1H)-酮(1a)与单取代BCB(2a)的作为模型反应。通过对波长的筛选发现,在400 nm下可基本抑制底物1a与2a的背景反应。基于此结果,研究者进一步对BpAD的结构进行修饰,合成了两种吸收光谱红移的辅因子MeO-BpAD和NMe₂-BpAD。在400 nm光照下,对一系列NAD⁺依赖型酮还原酶的野生型及突变体进行筛选。结果表明,当与工程化蛋白骨架Rr12aHSDH-M3结合时,MeO-BpAD可获得最佳反应结果(产率75%,92% ee)。NMe₂-BpAD因三线态能量与底物不匹配而表现出无活性。进一步引入A92Q突变,得到Rr12aHSDH-M4,反应结果进一步提升至77%产率和94% ee(图2)。
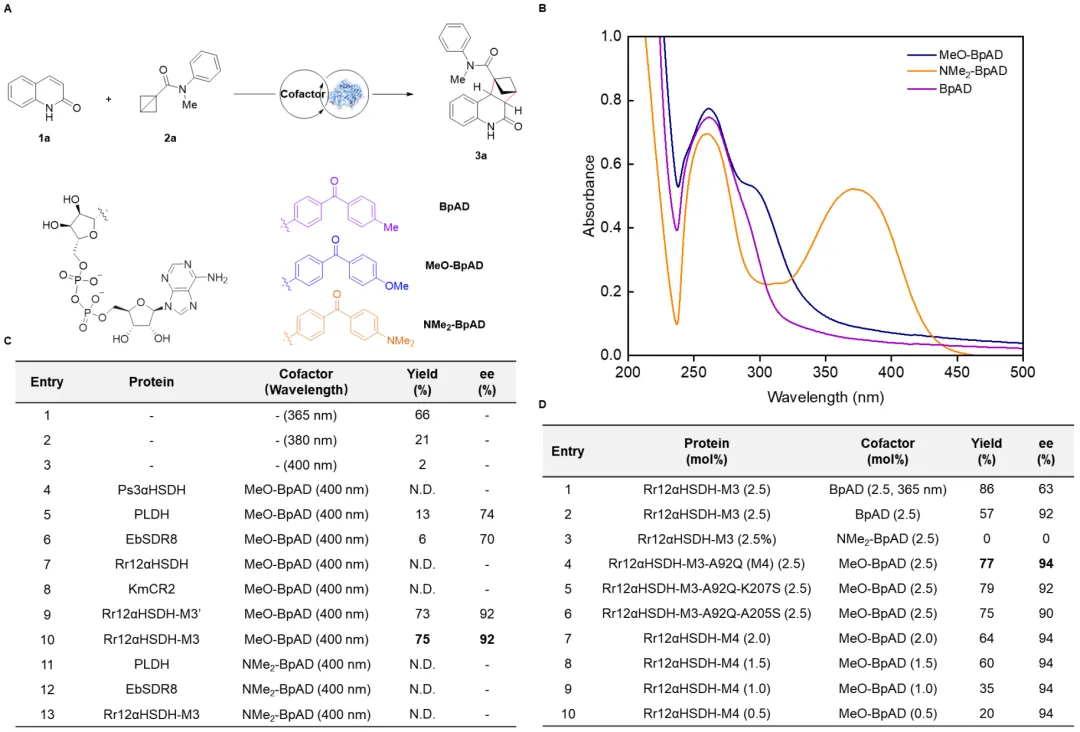
图2. 蛋白骨架的筛选和蛋白质工程(图片来源:J. Am. Chem. Soc.)
在最优反应条件下,该人工光酶体系具有较好的底物适用性,尤其对单取代联环丁烷(BCB)底物耐受性较好。带有不同位阻酰胺(从甲基到叔丁基,3a-3d)或不同链长酯基(3e-3h)的BCB均能以高产率(最高88%)和高对映选择性(高达94% ee)得到相应手性BCH产物。酰基取代的BCB(3i)也能有很好的兼容性。相比之下,对喹啉-2(1H)-酮(1)的修饰容忍度较低:喹啉酮的6-位取代会导致产率大幅下降(3k-3n),8-位取代进一步降低反应性(3o),1-位取代同时降低产率和对映选择性(3p),2-位取代则完全抑制产物生成(3q, 3r),N烷基取代也无法发生反应(3s),喹喔啉-2(1H)-酮类底物(如3t)表现出更高的反应性,但对映选择性有所降低。后续计算研究表明,这是由于蛋白活性口袋对喹啉酮的结合空间狭窄,微小的取代基位阻会破坏其结合取向,使反应双键难以与另一底物接触,而BCBs的结合区域空间充足,因此展现出更广的取代基耐受性,同时该反应体系可成功放大至0.34 mmol规模。
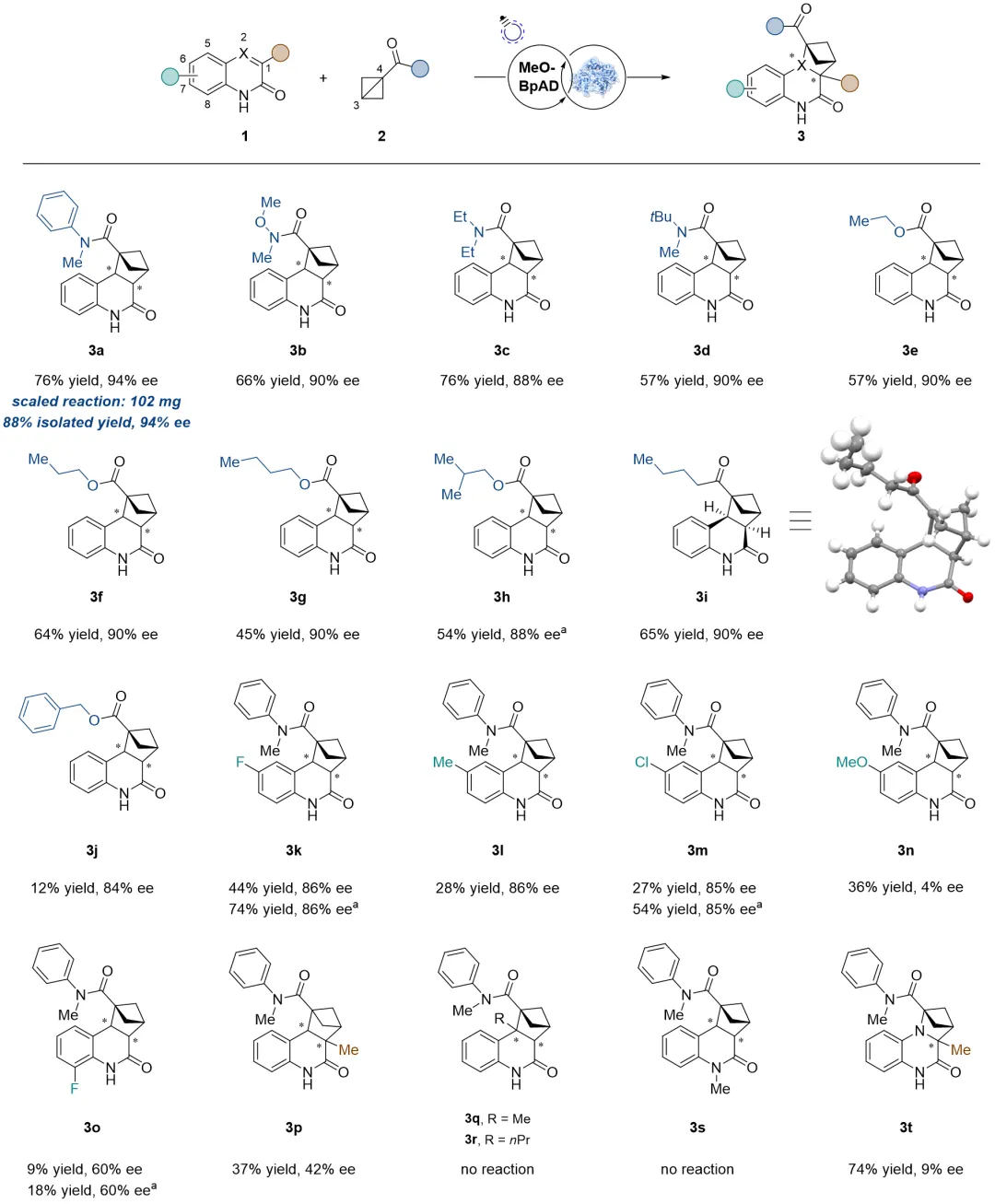
图3. 底物适用性(图片来源:J. Am. Chem. Soc.)
机理研究结合了实验与理论计算:等温滴定量热法测定表明结构修饰后的辅因子MeO-BpAD与蛋白质骨架(Rr12aHSDH-M4)仍可以保持一定结合力(Kd~2.3 μM)。瞬态吸收光谱显示,蛋白质的微环境使MeO-BpAD激发态寿命延长了约100倍(从0.66 μs提升至63 μs),增强了其能量转移能力。计算研究表明,反应遵循能量转移机制:MeO-BpAD吸收光后经系间窜越至三线态,并通过Dexter机制将能量转移给底物1a,使其成为三线态,随后三重激发态1a可激活2a形成激基复合物中间体3RC(exciplex),随后的C1-C3键形成是决速步。非共价相互作用(IGMH)分析和能量计算表明,在优势对映体(Re面进攻)的过渡态(³TS1Re)中,MeO-BpAD与1a之间存在显著的π-π堆积作用,且其静电相互作用能远低于非优势路径(Si面进攻,³TS1Si),这2.5 kcal·mol⁻¹的能垒差是立体选择性的主要来源。计算还解释了喹啉酮类底物范围受限的原因:底物1a的结合腔空间狭窄,因此在底物1上的取代基会通过空间位阻破坏1与2a的最佳反应几何构型。
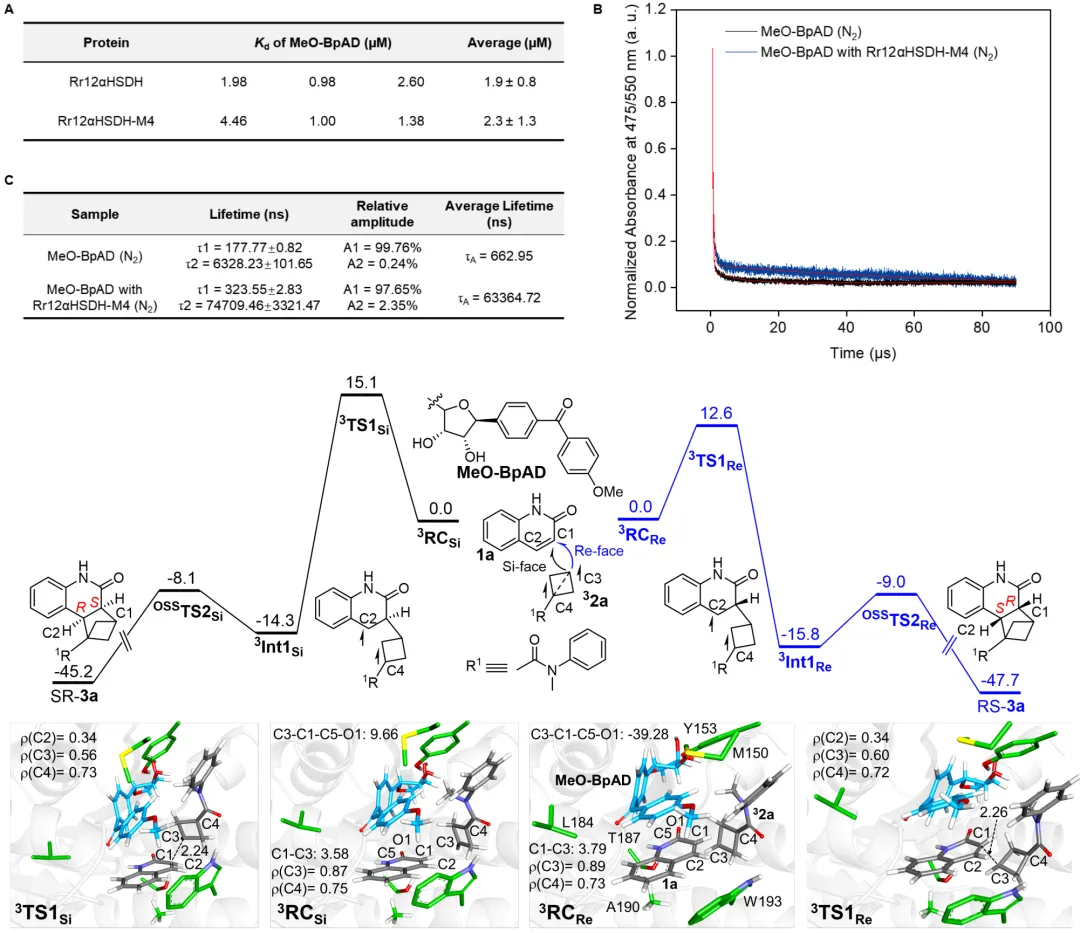
图4. 机理探究(图片来源:J. Am. Chem. Soc.)
本研究通过合理设计并引入甲氧基修饰,开发了新型光活性辅因子MeO-BpAD,进而构建了一种高效的人工光酶催化系统。该系统成功实现了喹啉-2(1H)-酮与单取代联环丁烷之间的对映选择性分子间[2π+2σ]环加成反应,为合成具有重要价值的手性双环[2.1.1]己烷生物电子等排体提供了一条新途径。这项工作充分展示了一种高度模块化的策略:通过理性设计光活性辅因子和蛋白质工程,实现光化学反应的立体控制。相比传统光催化体系,这种人工光酶不仅能够抑制背景反应,还能在分子层面精确调控反应路径。更重要的是,通过设计可调控的光活性辅因子,将天然酶“重编程”为具有全新反应类型的催化剂。这种思路有望进一步拓展至更多困难的自由基反应与环加成体系,为复杂分子的不对称合成提供全新的解决路径。
这一成果近期发表在Journal of the American Chemical Society上(DOI:10.1021/jacs.5c20918),本文的理论计算部分由厦门大学王斌举课题组完成,其余部分由南京大学潘惠杰课题组完成。南京大学21级博士研究生李劲思、22级博士研究生杜平和厦门大学22级博士研究生周太平为论文的共同第一作者。南京大学潘惠杰特聘研究员和厦门大学王斌举教授为论文的共同通讯作者。
论文信息
Enantioselective [2π+2σ] Cycloaddition to Bicyclo[2.1.1]hexanes Enabled by an Artificial Photoenzyme
Jinsi Li, $ Ping Du, $ Tai-Ping Zhou, $ Wenhao Hu, Haoyu Li, Binju Wang, * and Hui-Jie Pan*
https://doi.org/10.1021/jacs.5c20918



| 点击即可阅读合集 | ||
| 催化化学 | ||
| 分析化学 | ||
| 生物化学 |

随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- 沂州东院丨南京路上第四代墅厅大平层,临沂好房子的进化方向
- 眼周嘴角反复干痒?警惕神经性皮炎_南京哪家医院看神经性皮炎最好
- 济南没想到,南京也没想到,如今的中国徐州,已成为南北差异化视角下的独特焦点!
- 南京,1994命案
- 南京工业职业技术大学2026年高层次人才招聘公告
- 【国内团】4月18日丨【考察团】南京宜兴长兴双高5日游
- J.Cole南京同曦 , 当你有实力,追星变得如此简单..还剩9场球可以看到JCole
- 春假遇清明,健康不放假!南京中小学生“五健”守护指南
- 南京农业大学教授入选国家高层次青年人才、以通讯作者在《Nature》子刊发表研究论文,揭示稻曲病菌“一箭双雕”致病新机制
- 【JACS】西北工业大学谷龙&南京工业大学安众福/马会利&新加坡南洋理工大学Yaru Gao|有机X射线闪烁体中用于动态成像的快速三重激子利用
