南京大学周勇/东南大学李强,最新JACS!设计独特非晶FeOₓ介孔纳米片实现结构-电子协同作用助力高选择性/高效光转化CO₂制多碳燃料!
将CO₂光催化转化为高能量密度碳氢化合物面临重大挑战,不仅需要高光子利用效率,还需要优异的催化活性。
2026年04月04日,邹志刚院士、南京大学周勇、东南大学李强团队合作在Journal of the American Chemical Society期刊发表题为“Design of Unique Amorphous FeOx Mesoporous Nanosheets for Structural-Electronic Synergy toward Highly Selective and Efficient CO2 Photoconversion into Multicarbon Fuels”的研究论文,南京大学Cheng Tingting、东南大学Lin Wangqiang为论文共同第一作者,周勇、李强为论文共同通讯作者。
第一作者:Cheng Tingting、Lin Wangqiang
通讯作者:周勇、李强
通讯单位:南京大学、东南大学
论文DOI:10.1021/jacs.6c01273
该研究提出了一种合理设计的光催化剂,利用非晶FeOₓ的结构灵活性和可调电子特性来有效满足这些要求。该催化剂基于多个协同因素,在520 nm处表现出1.60%的表观量子效率,乙烷选择性约为70%(基于产率)和约94%(基于电子)。具体而言,非晶骨架结合介孔结构提供了丰富的无序位点,极大地增强了分子吸附和活化;介孔薄壁缩短了电荷向表面的扩散路径;激光诱导的氧空位提高了Fe²⁺/Fe³⁺比例,并建立了一个串联催化路径,其中Fe²⁺可以通过稳定COCO中间体有效促进C=C偶联。该研究为通过跨尺度结构协同作用将CO₂转化转向高附加值多碳燃料提供了设计原则。
利用太阳能等可再生能源将CO₂转化为高可用性的清洁燃料而非传统燃料,已成为碳回收和碳中和最有前景的方法之一。然而,由于C=O键的高稳定性,光催化CO₂还原仍然是一项重大挑战,当前体系主要生成C₁产物,如CO、CH₄和CH₃OH。尽管已经取得了进展,包括该研究团队开发了用于乙烯(C₂H₄)的原子薄层AgInP₂S₆纳米片以及用于丙烷(C₃H₈)的Cu单原子锚定Ti₀.₉₁O₂原子薄层单层等光催化剂,但高效、选择性地生成高价值多碳(C₂₊)碳氢化合物仍然受到几个关键瓶颈的阻碍,包括足够的e⁻/h⁺稳定中间体、合适C=C偶联能垒以及维持有效电荷分离和传输。
为解决这些长期存在的难题,有前景的光催化剂的合理设计需要同时优化原子尺度的无序性和纳米尺度的孔隙率。非晶材料,顾名思义,缺乏长程原子序,导致与其晶态对应物相比,缺陷和不饱和配位位点的密度增加。在非晶催化剂中调控原子结构通常为调节电子性质和化学环境提供了有前景的机会,这可以显著提高水分解、CO₂还原以及有机分子光催化氧化中的光催化性能。同时,介孔材料的特点是具有丰富的相互连接的孔道,这显著增加了表面积与体积比,为吸附分子与催化剂相互作用提供了更多可及的活性位点。为了有效整合这些结构优势,FeOₓ因其地球丰度高、优异的化学稳定性、可调铁氧化态(Fe²⁺/Fe³⁺)以及适应性强的缺陷化学性质而成为理想平台。这些固有性质使FeOₓ成为一个多功能基础,用于精确调控电荷传输、能带对齐和催化反应性,正如在各种催化体系中所证明的那样。
在此,该研究成功制备了非晶FeOₓ介孔纳米片(命名为FeOₓ-ns),随后通过脉冲激光刻蚀处理,同时增加了氧空位(Vₒ)浓度和Fe²⁺/Fe³⁺比例。这种结构与电子修饰的结合导致光活性显著提高,并进一步生成深度反应产物乙烷(C₂H₆),其基于产物的选择性约为70%,基于电子的选择性约为94%,在520 nm处表观量子效率(AQE)达到1.60%。所获得的高效率与高选择性源于非晶无序、介孔结构以及Fe²⁺/Fe³⁺氧化还原化学之间的集体协同作用。无序结构提供了丰富的不饱和配位位点,有利于CO₂的吸附与活化;而介孔网络则最大化了可及活性位点,减轻了扩散限制,加速了电荷向表面的传输,并提供了有利于C−C偶联的空间限域效应。同时,Fe²⁺与Fe³⁺物种在空间上的紧密耦合实现了一种串联催化机制:CO₂活化和*CO中间体的生成发生在Fe³⁺位点上,随后在相邻的Fe²⁺位点上进行C−C偶联,从而有效抑制了*CO的解吸,并因此稳定了*OCCO中间体。该研究为光催化CO₂还原提供了一个通用设计原则,展示了如何利用结构与电子的集体协同作用,选择性地驱动C−C偶联以生成高价值的多碳产物。
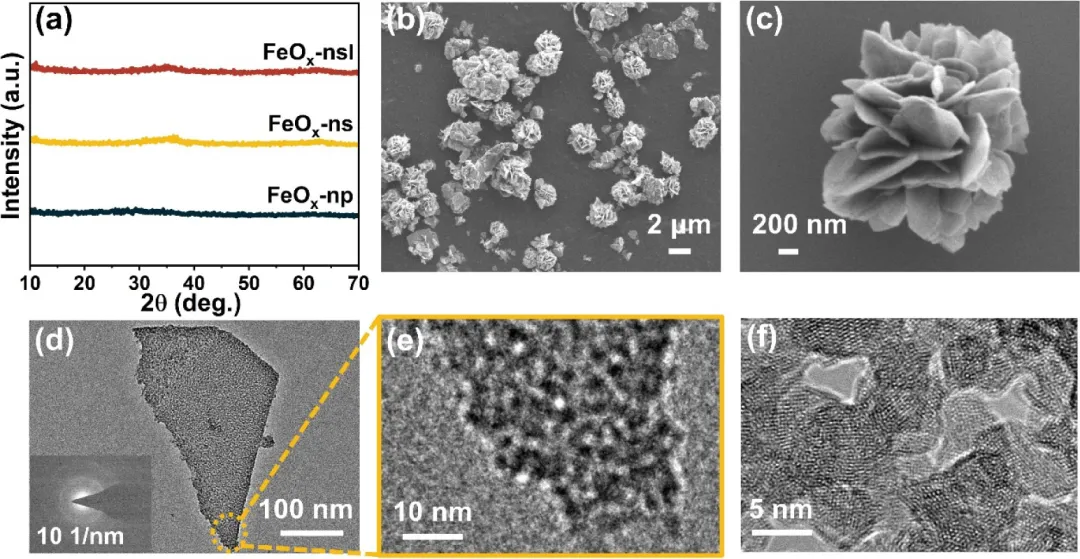
图1:(a) FeOₓ的XRD谱图,(b-c) SEM图像,(d-f) TEM图像(插图:SAED图)以及FeOₓ-ns的HRTEM图像。
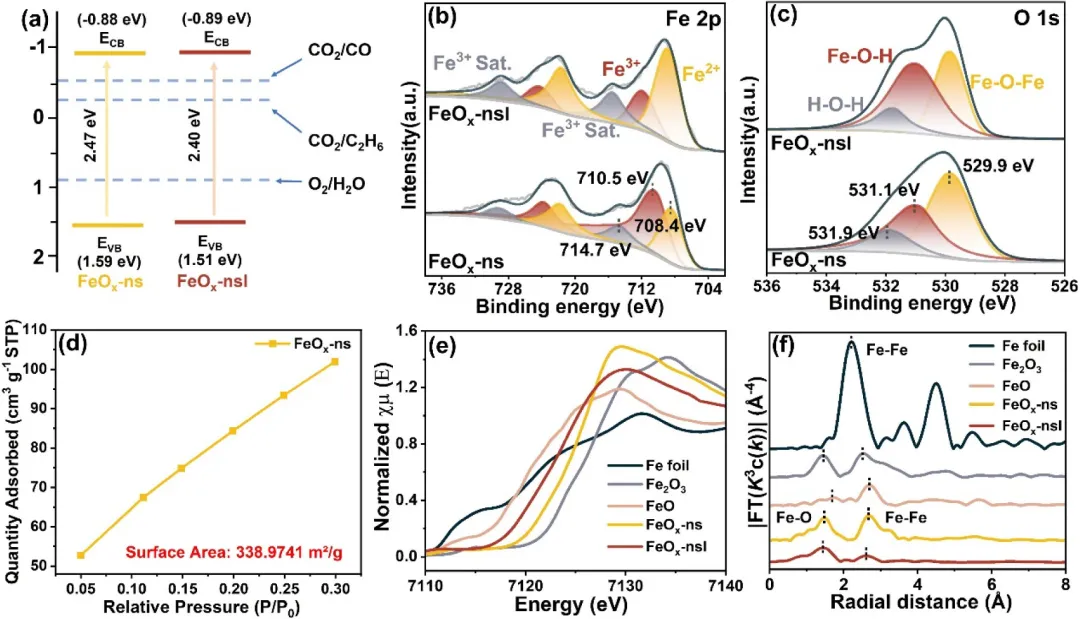
图2:(a) 能带位置图,(b) 样品的Fe 2p和(c) O 1s的XPS谱图,(d) FeO-ns的氮气吸附等温线,(e) 样品的Fe K边XANES谱图和(f) Fe K边EXAFS谱图的傅里叶变换。
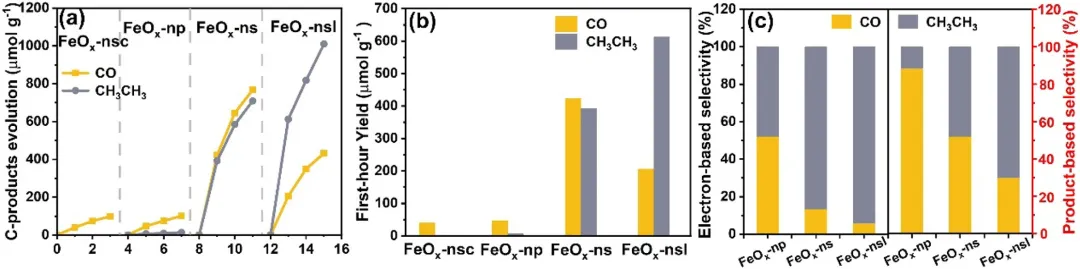
图3:(a) 含碳产物演化曲线;(b) FeO-nsc、FeO-np、FeO-ns和FeO-nsl第一个小时的光催化活性比较;(c) FeO-np、FeO-ns和FeO-nsl的基于电子的选择性和基于产物的选择性。
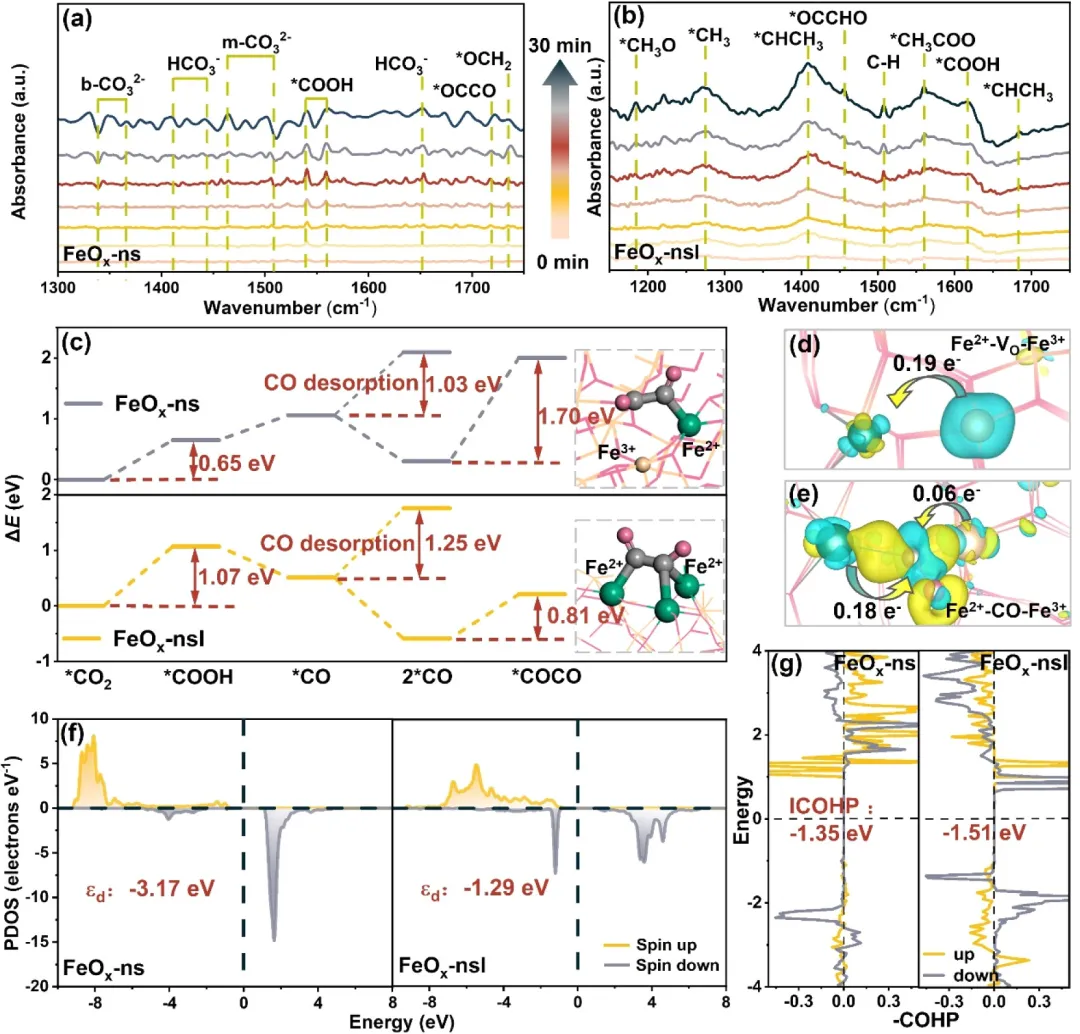
图4:(a)FeOₓ-ns和(b) FeOₓ-nsl上光催化CO₂还原的原位DRIFTS谱图。(c) FeOₓ-ns和FeOₓ-nsl上CO₂还原为COCO的反应能量变化,插图为相应的原子结构配置插图。(d) Fe²⁺-Vₒ-Fe³⁺和(e) Fe²⁺-CO-Fe³⁺结构的电荷密度差(黄色代表电子积累,蓝色代表电子耗尽)。(f) FeOₓ-ns和FeOₓ-nsl的PDOS和d带中心。(g) FeOₓ-ns和FeOₓ-nsl的ICOHP。
总之,该研究成功合成了非晶FeOₓ介孔纳米片,并通过脉冲激光刻蚀增加了氧空位浓度,从而提高了Fe²⁺/Fe³⁺比例。这种对几何结构和电子结构的双重调控将CO₂光还原路径转向选择性地生成乙烷,实现了约70%的基于产物的选择性、约94%的基于电子的选择性以及在520 nm处1.60%的AQE。高达24.42 cm³g⁻¹的CO₂吸附容量与其结构特性相关,包括非晶性质、介孔性和富含缺陷的表面。通过BET和原位DRIFTS分析阐明了介孔网络在增强质量传输和提供空间限域方面的作用。理论计算进一步揭示了一种串联催化机制,其中Fe³⁺主导CO的产生,而相邻的Fe²⁺通过稳定*COCO中间体促进C-C偶联,有效抑制了CO的解吸。该研究展示了一种整合结构无序、纳米级孔隙率和定制氧化还原化学的合理设计原则,以共同驱动选择性将CO₂转化为高附加值多碳燃料。