COFs是一种由有机单元通过共价键构筑的多孔晶态材料,在气体储存、分离及催化等领域具有重要潜力,但其发展受到两大核心问题的限制:一是由于动态共价键形成过程存在动力学障碍,难以获得高结晶度结构,从而影响材料的孔隙性与稳定性;二是传统合成通常依赖大量有机溶剂(如间三甲苯、NMP等),不仅不利于规模化生产,也不符合绿色可持续发展的需求。此外,由于多数单体具有疏水性,在水中溶解度极低,使得实现纯水体系下的COFs合成极具挑战性。因此,如何在绿色溶剂(水)中实现高结晶度、高孔隙度COFs的可控构筑,并突破单体溶解性差与反应可逆性不足等问题,成为该领域亟待解决的关键科学问题。
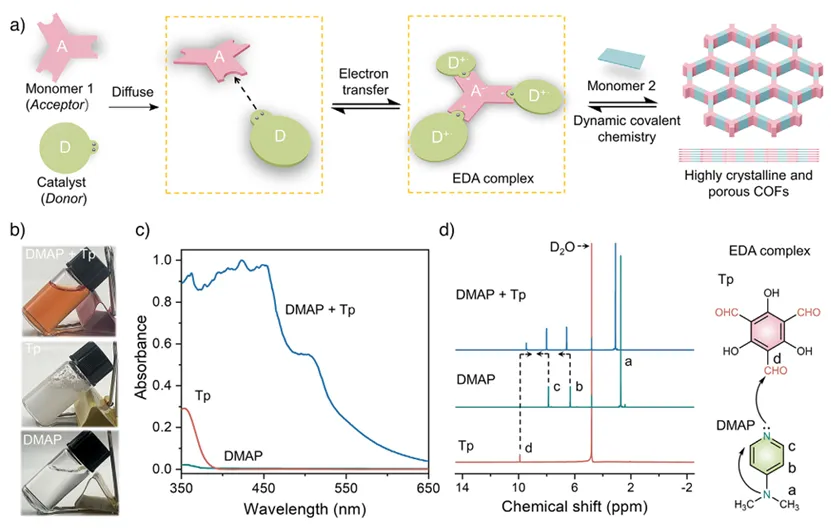
首先,作者系统展示了电子给体–受体(EDA)相互作用在水相中促进COFs合成的形成过程及其多维度实验证据。在示意图(图1a)中,作者提出电子缺陷的醛类单体(如Tp)可与电子富集的催化剂DMAP通过分子间电子转移形成EDA复合物,这一过程能够在反应初期实现单体的预组织,从而为后续有序的动态共价组装奠定基础。随后,通过直观的实验照片(图1b)可以看到,原本在水中呈浑浊分散状态的Tp,在加入DMAP后迅速转变为均一透明的橙色溶液,这一显著变化表明EDA复合物的形成不仅改变了体系的光学性质,也显著提升了疏水单体在水中的溶解性。进一步地,紫外-可见吸收光谱(图1c)中出现明显的吸收红移,说明体系中发生了有效的电子转移过程,这是EDA相互作用的典型特征;而¹H NMR谱图(图1d)中DMAP与Tp相关氢信号分别发生高场和低场位移,则从分子层面进一步验证了电子从DMAP向Tp转移,导致电子云分布发生改变。
图1. Tp与DMAP在水中形成EDA复合物的表征。(a)电子受体单体与电子给体催化剂之间形成EDA复合物用于COFs合成的示意图;(b) 0.15 mmol Tp、1.8 mmol DMAP及其在4 mL水中形成的EDA复合物的对应照片;(c)Tp、DMAP及其EDA复合物的归一化紫外–可见吸收光谱(所有光谱均在水中、使用1 mm光程石英比色皿测定);(d)Tp、DMAP及其EDA复合物(DMAP:Tp摩尔比为3:1)在D₂O中的¹H NMR谱图以及EDA复合物的化学结构。
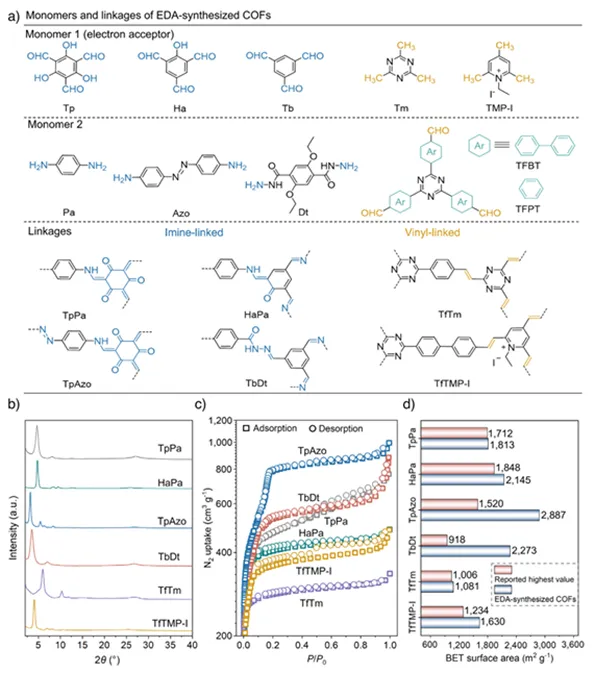
接着,作者从结构多样性、结晶性以及孔结构性能三个方面系统展示了基于EDA策略合成COFs的优越性。首先,在(图2a)中给出了不同单体及其构筑的COFs结构示意,说明该方法不仅适用于亚胺键连接体系,还可拓展至乙烯基等多种连接类型,体现出良好的普适性。粉末X射线衍射图谱(图2b)显示所有材料均具有清晰且尖锐的衍射峰,表明其具有高度有序的晶体结构。高分辨透射电镜结果也进一步佐证了这一点。氮气吸附–脱附等温线(图2c)则揭示这些COFs均具有典型的多孔特征和较高的比表面积。更为重要的是,作者对比了本工作与已报道COFs的BET比表面积,可以看到EDA策略所得材料的比表面积接近甚至达到理论极限,刷新了多种COFs体系的最高纪录(图2d)。这表明通过EDA相互作用提升反应可逆性与单体溶解性,有效减少了结构缺陷,使框架更加规整有序。
图2.不同单体构筑的EDA合成COFs的结晶性与孔结构表征。(a)单体的化学结构及EDA策略合成的COFs结构片段;(b) EDA合成的TpPa(灰色)、HaPa(绿色)、TpAzo(蓝色)、TbDt(红色)、TfTm(紫色)和TfTMP-I(金黄色)的PXRD图谱;(c)在77 K下测得的氮气吸附(方块)与脱附(圆点)等温线;(d)EDA合成COFs(蓝色柱)与已报道具有最高BET比表面积的COFs(红色柱)的对比。
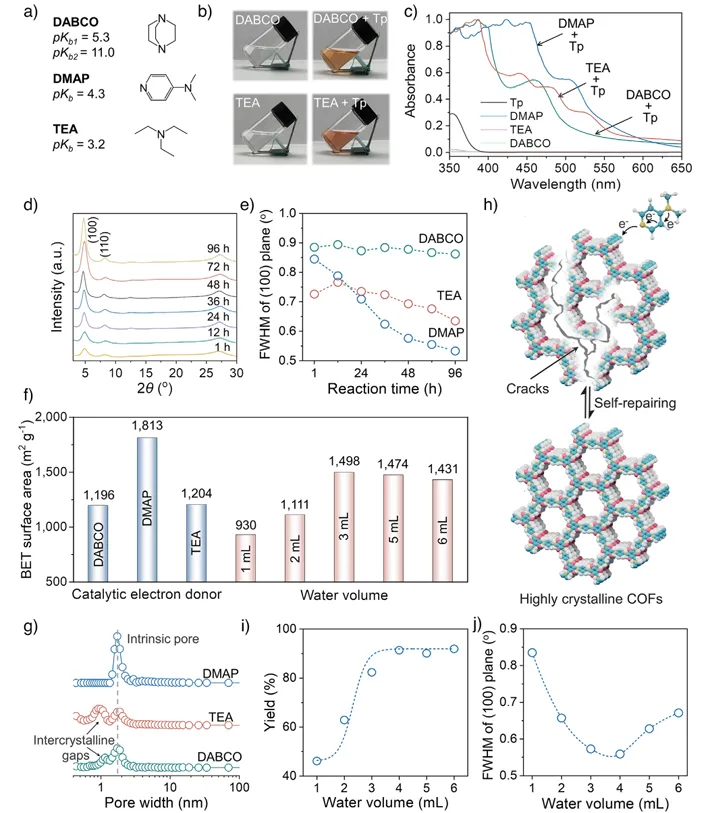
作者还系统探讨了催化电子给体种类及水用量对EDA策略合成COFs性能的影响机制。首先,作者展示了三种不同电子给体催化剂(TEA、DABCO和DMAP)的结构及其碱性差异(图3a),图3b中相应的实验照片表明它们均可与Tp形成EDA复合物并提升其在水中的溶解性,但颜色变化程度不同,暗示相互作用强弱存在差异。紫外–可见光谱进一步证实DMAP体系具有最明显的红移,说明其电子给体能力最强(图3c)。随后,作者通过PXRD及半峰宽变化揭示,随反应时间延长,晶体逐步完善,而DMAP体系表现出最高结晶度(图3d-e)。图3f-g中的BET比表面积及孔径分布结果表明,DMAP催化下所得材料孔结构最规整、比表面积最大,而TEA和DABCO体系中出现额外孔隙,说明缺陷较多。图3h说明了EDA作用增强反应可逆性,从而实现“自修复”形成高有序结构。最后,作者考察了水用量影响,结果表明适量水可提高产率和结晶度,但过多则因EDA复合物浓度降低而不利于结构形成(图3i-j)。
图3. 不同催化电子给体及水用量对TpPa的EDA合成影响。(a)三种电子给体催化剂的化学结构及pKb值;(b) DABCO、TEA及其EDA复合物在水中的照片;(c)不同电子给体形成的EDA复合物的归一化紫外–可见吸收光谱(均在水中、使用1 mm光程石英比色皿测定);(d)采用DMAP作为催化电子给体,在不同反应时间下合成TpPa的PXRD图谱;(e)对应(100)晶面半峰宽(FWHM)的变化;(f)不同电子给体条件下以及不同水用量条件下合成TpPa的BET比表面积;(g)不同电子给体条件下TpPa的孔径分布;(h)通过自修复过程形成高结晶COFs的示意图;(i)不同水用量下TpPa的产率;(j)在DMAP催化下,不同水用量合成TpPa的(100)晶面半峰宽变化。
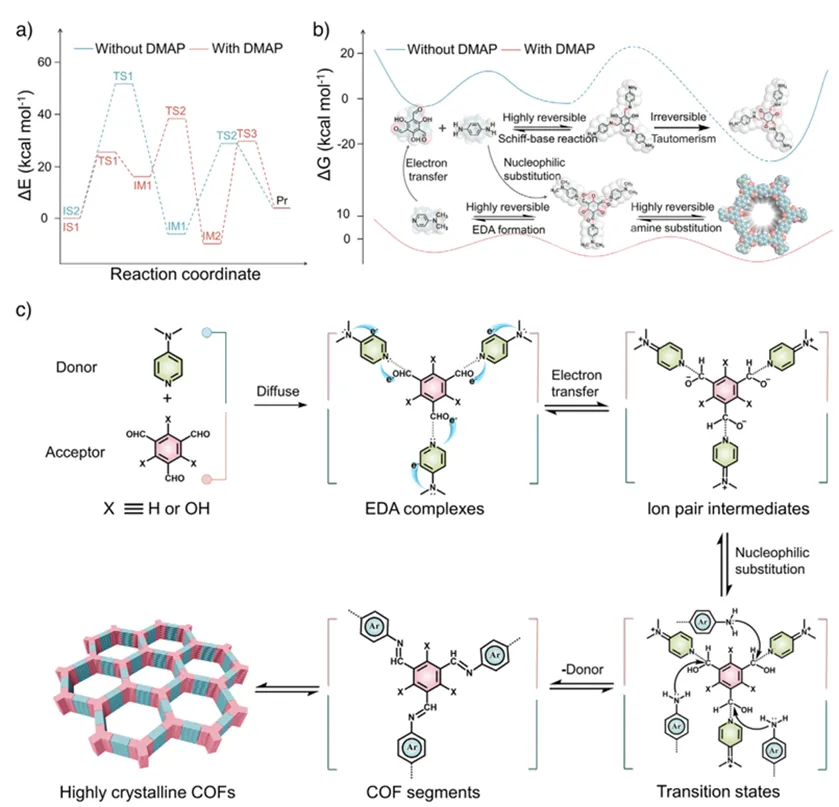
随后,作者从理论计算角度深入揭示了EDA策略促进COFs形成的反应机理与能量变化过程。首先,作者通过密度泛函理论(DFT)计算比较了有无DMAP参与时Tp与Pa反应的能量路径(图4a)。在未引入DMAP时,反应需经历较高的活化能才能发生缩合,而在存在DMAP时,其孤对电子首先诱导向Tp的电子转移,形成EDA复合物中间体,从而显著降低后续亲核取代反应的能垒,使反应更易进行。接着,作者还通过吉布斯自由能变化对比表明,传统直接缩合路径由于包含不可逆的互变异构步骤,整体能量需求较高;而经由EDA复合物的路径则能同步实现电子转移与键构筑过程,降低能耗并增强反应可逆性,有利于错误结构的自修复,从而提升结晶度(图4b)。最后,作者总结了基于EDA机制的反应路径:电子给体(如DMAP)与电子受体单体形成复合物并提高其水溶性,随后被第二单体取代完成框架构筑(图4c)。
图4. 基于EDA策略合成TpPa的反应机理与路径分析。(a) Tp与不同电子给体反应的能量变化路径,其中Tp与Pa在有无DMAP条件下的混合体系被设定为初始态(IS),最终生成TpPa产物(Pr);(b)通过可逆席夫碱反应及不可逆β-酮烯胺互变异构的直接缩合路径,与通过EDA复合物形成路径的吉布斯自由能变化对比;(c)经由EDA复合物进行反应的路径示意及分子在分子间电子转移过程中的结构转变。
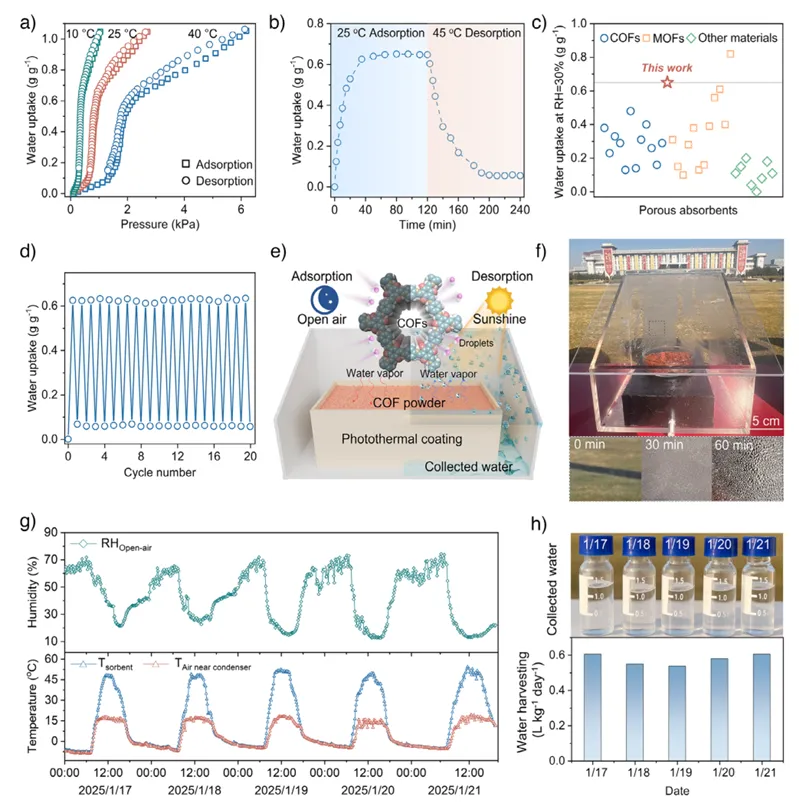
最后,作者展示了基于EDA策略合成的TpPa在空气取水方面的性能及实际应用潜力。作者测试了不同温度下的水蒸气吸附等温线,可以看到材料在低湿度区(约20%–30% RH)就出现陡峭的吸附阶跃,说明其对水分子具有很强的亲和能力,尤其适用于干旱环境(图5a).动态吸附–脱附曲线进一步表明,该材料能够在较短时间内快速吸水并在适度升温下释放水分,体现出良好的动力学性能与可再生性(图5b)。作者还将其与其他多孔材料进行对比,可以发现该COF在低湿度条件下的吸水能力处于领先水平,突出其结构优势(图5c)。循环测试结果显示材料在多次吸附–脱附过程中性能几乎无衰减,证明其具有优异的稳定性(图5d)。进一步地,作者展示了实际构建的空气取水装置及其运行情况,通过昼夜温湿度变化实现吸附与释放过程(图5e-f)。作者还记录了装置运行过程中环境与材料温湿度变化(图5g)。最后作者给出了实际集水速率及收集到的水样照片(图5h)。
图5. EDA合成的TpPa用于空气取水的性能。(a)在10 ℃(绿色)、25 ℃(红色)和40 ℃(蓝色)下测得的水蒸气吸附等温线;(b)在30%相对湿度下,25 ℃吸附和45 ℃脱附的动态水吸附–脱附曲线;(c)不同多孔材料在空气中水吸附能力的对比(详见表S3);(d)吸附(25 ℃)–脱附(45 ℃)循环稳定性测试;(e)空气取水装置的设计示意图;(f)装置实物照片及冷凝器壁面随时间形成的水滴;(g)露天环境中空气、冷凝器附近空气及TpPa粉末的温度与湿度变化记录;(h)空气取水速率及对应收集水的照片(测试地点为南京东南大学操场)。
综上所述,该文章提出了一种基于电子给体–受体相互作用的全新策略,实现了共价有机框架在纯水体系中的高效合成。作者通过引入电子富集的催化剂(如DMAP)与电子缺陷单体形成EDA复合物,不仅显著提高了疏水单体在水中的溶解性,还增强了反应的可逆性,从而有效降低能垒、促进“自修复”过程,最终获得具有接近理论极限比表面积和极高结晶度的COFs材料。该方法具有良好的普适性和可扩展性,可适用于多种键型并实现规模化制备。基于此策略构筑的COFs在空气取水方面表现出优异性能,在低湿度条件下仍具较高吸水能力,并能稳定循环使用,展示出在可持续水资源获取等领域的重要应用潜力。
Title:Electron Donor–Acceptor Interactions Enabling Aqueous Synthesis of Covalent Organic Frameworks with Unprecedented Crystallinity and Porosity for Water Extraction from Arid Air
Authors: Jinglin Gao+, Congcong Yin+, Meng Chen, Mingjie Wei, Chuansheng Tang, Ya Du, Xin Zhao, Yuping Wu, and Yong Wang*
To be cited as: Angew. Chem. Int. Ed., 2026, 65, e15651.
DOI: 10.1002/anie.202515651
研究方向|超分子化学
Bigger Deeper Smarter Higher
投稿,荐稿,合作
437015451@qq.com