「上海岱算科技有限公司」已向境内外230余家高等院校/科研院所提供了累计1400多项模拟计算服务,赋能科学研究提速增效!合作实验课题组在线发表学术论文期刊有ACS系列、AM系列、Angew、CEJ、EST、JACS、Matter、Nature子刊等,助力科研工作锦上添花!在高浓度生物质平台分子的电催化加氢(ECH)过程中,性能下降仍然是实际应用面临的主要障碍,这主要是由于对浓度依赖机制的理解尚不完整。
2026年05月04日,南京林业大学荆宇团队在Angewandte Chemie International Edition期刊发表题为“Circumventing Concentration Limitations in Electrocatalytic Hydrogenation of 5-Hydroxymethylfurfural through Alkali Metal Ion Mediated Supramolecular Control”的研究论文,团队成员刘天阳、杨有超为论文共同第一作者,荆宇为论文通讯作者。
第一作者:刘天阳、杨有超
通讯作者:荆宇
通讯单位:南京林业大学
论文DOI:10.1002/anie.3764137
该研究以5-羟甲基糠醛还原反应(HMFRR)作为模型体系,通过恒电势密度泛函理论DFT计算和从头算分子动力学AIMD模拟,揭示了在高反应物浓度下分子间氢键的一个出乎意料的作用。在这一机理认识的指导下,开发了一种阳离子调控策略,并发现Li⁺离子是HMFRR最有效的促进剂,其协同作用包括:(i) 由于Li⁺离子半径小、能深入内亥姆霍兹平面且具有低配位结构,从而破坏HMF二聚体中的分子间氢键;(ii) 因其强吸电子特性而增强的*H转移;(iii) 在中等Li⁺浓度下保持高的HMFRR选择性。实验电化学测量和¹H核磁共振(¹H NMR)光谱验证了这些预测。该研究解决了高浓度电催化加氢中一个长期存在的挑战,并建立了一个通过合理界面工程升级浓缩生物质衍生平台分子的通用范式。
电催化加氢(ECH)作为一种可持续且低排放的策略,在温和条件下对低价值或污染物(如CO₂和NO₂)进行增值转化,正受到越来越多的关注。它也为将生物质衍生的平台分子升级为有价值的化学品提供了一条有前景的途径。尽管有这些优势,但由于其转化速率有限、操作成本高以及可扩展性差,该技术仍远未取代传统热催化过程。核心挑战在于实现具有工业相关性的、高浓度的电催化加氢过程,该过程既能通过减少废物和下游分离成本来提供高通量,又能改善经济效益。
高浓度反应物下的电合成通常会遭受严重的性能下降,从而阻碍大规模实施。固有的传质限制、竞争性副反应、电极污染和钝化以及高浓度条件下离子迁移率降低已被广泛认为是影响因素。为了克服这些问题,已经开发了各种策略来增强传质,例如使用流动池、微流体反应器和气体扩散电极以实现更高的反应物利用率。然而,一个同样关键的因素在很大程度上被忽视了,即高浓度下反应物之间的分子相互作用对内在电催化活性的影响。因此,全面理解高浓度条件下的电催化加氢机制对于设计既能实现高效率和经济效益的电合成有效策略至关重要。
这一挑战对于生物质衍生的平台分子尤为突出,其结构复杂性促进了广泛的分子间相互作用以及在电催化加氢过程中与反应中间体的交叉反应,导致在浓缩条件下发生催化自中毒。尽管它们具有工业重要性,但这种浓度依赖的性能下降的机理起源仍然知之甚少,这凸显了对详细分子水平认识的迫切需要。一个具有代表性的体系是5-羟甲基糠醛(HMF),它是异构化的C6糖脱水产生的一个关键中间体,并被美国能源部认定为十大最有前景的生物质衍生平台分子之一。2024年全球HMF市场估值约为6330万美元,预计到2033年将达到7390万美元。电化学HMF还原反应(HMFRR)提供了一条可持续的途径来生产高附加值化学品,包括2,5-二(羟甲基)呋喃(DHMF)、2,5-二甲基呋喃(DMF)和2,5-二(羟甲基)四氢呋喃(DHMTHF),这些产品对于能源、制药和化学应用至关重要。在这些可能的产物中,DHMF是合成功能化聚醚、聚酰胺和各种药物中间体的关键前体。虽然先前的大多数研究都考察了稀原料(通常为20 mM),但HMFRR的性能和DHMF的法拉第效率在较高浓度下通常会急剧下降,从而增加了产物分离成本。这就提出了一个关键问题:是什么基本因素抑制了高浓度下的HMFRR?此外,能否合理设计界面微环境以在高浓度条件下维持高效的ECH活性?
在此,该研究进行了最先进的恒电势密度泛函理论DFT计算和从头算分子动力学AIMD模拟,以阐明生物质衍生的HMF在高浓度下的ECH机理。以铜(Cu)催化剂上的HMFRR作为模型体系,确定分子间氢键(H-键)是控制反应路径的关键因素,并为电催化性能的浓度依赖性下降提供了机理解释。在这一机理认识的指导下,引入碱金属阳离子(Li⁺、Na⁺和K⁺离子)来调控界面微环境。其中,Li⁺通过破坏HMF二聚体之间的分子间H-键网络并促进*H物种转移,最有效地促进了HMFRR。理论预测通过¹H核磁共振(¹H NMR)分析和电化学表征进行了实验验证,为克服电催化加氢中浓度依赖性限制提供了统一的机理认识和通用策略。
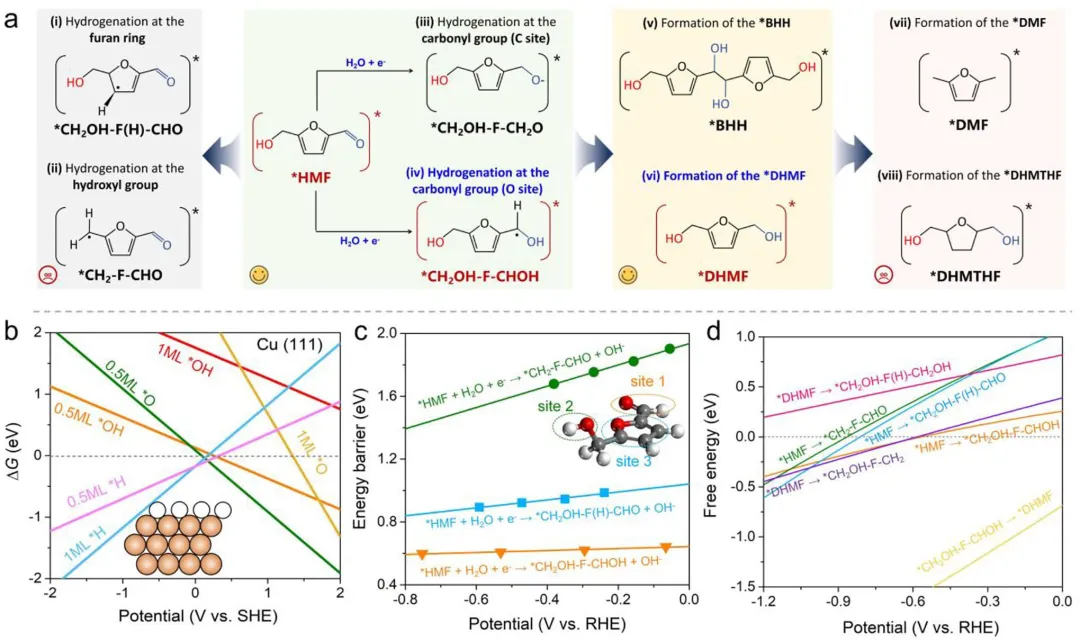
图1 (a) HMF电化学升级反应网络示意图。(b) 表面Pourbaix图以及pH=7时最有利的*H-Cu表面示意图。(c) 不同HMF加氢过程(包括醛基(位点1)、醇基(位点2)和呋喃环(位点3)上的加氢)的动力学能垒随电势变化的情况。(d) 较低浓度下HMFRR在*H-Cu(111)上的基元步骤自由能变化与电势的函数关系。
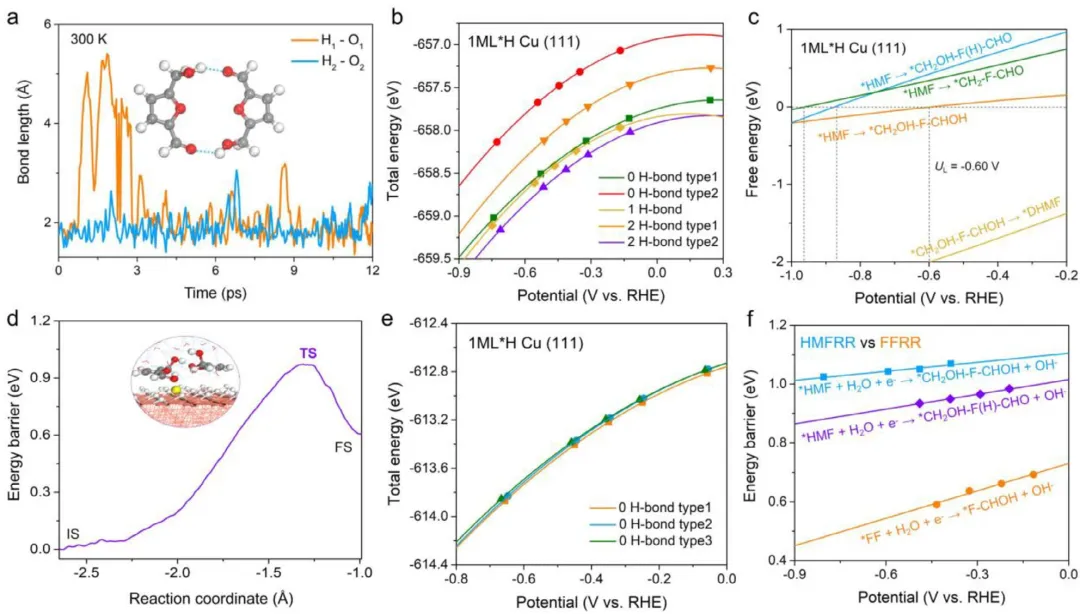
图2 (a) 在12 ps AIMD模拟过程中,HMF二聚体内氢键长度的时间演化。(b) 不同类型HMF二聚体在*H-Cu(111)上的总能量随电势变化的情况。(c) 在高浓度下,HMFRR在*H-Cu(111)上生成*DHMF的自由能变化与电势的函数关系。(d) 在高HMF浓度下,使用约束AIMD模拟获得的*HMF向*CH₂OH-F-CHOH氢化反应的动力学能垒图。插图显示了该过程的过渡态。(e) 两个FF分子在不同构型排列下的总能量随电势变化的情况。(f) *HMF和*FF在*H-Cu(111)上氢化过程的动力学能垒与电势的函数关系。
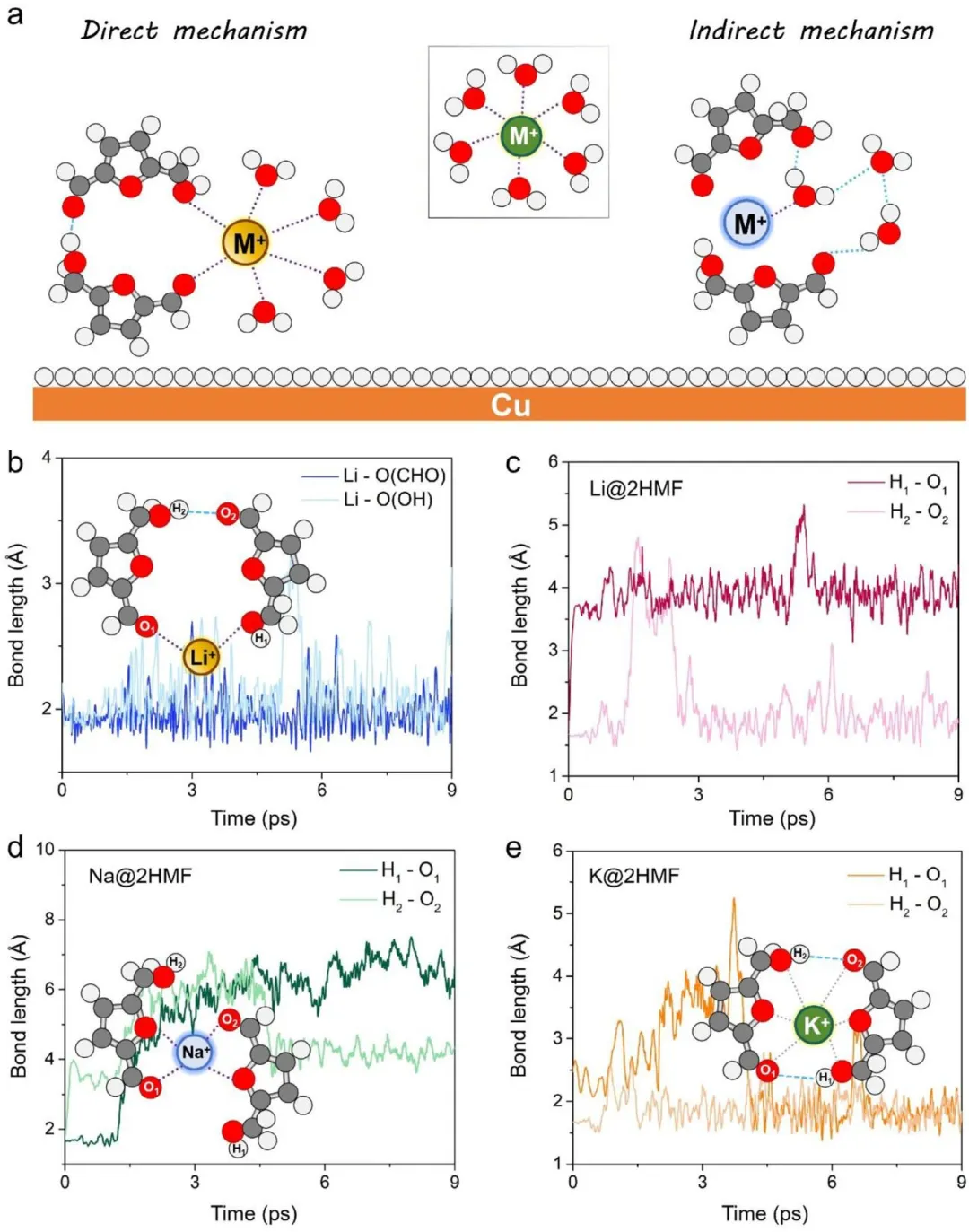
图3 (a) 碱金属阳离子破坏*H-Cu(111)上HMF二聚体构型及其溶剂化壳层的示意图。(b) 在300 K下9 ps AIMD模拟过程中Li-O键长的时间演化。(c-e) 在300 K下9 ps AIMD模拟过程中,(c) Li@2HMF、(d) Na@2HMF和(e) K@2HMF体系中氢键长度的时间演化。
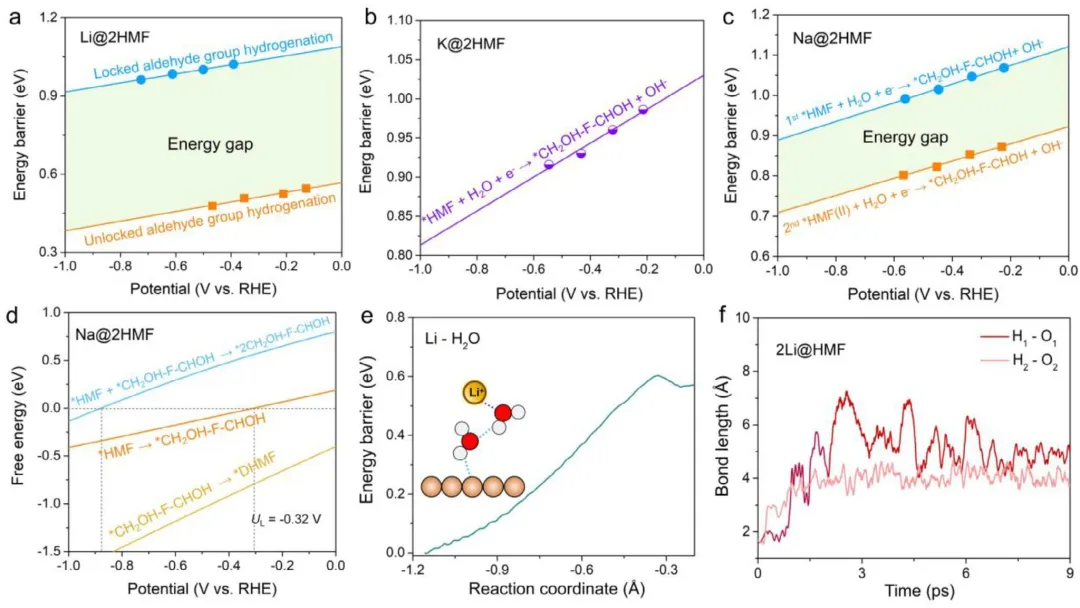
图4 (a) Li@2HMF、(b) K@2HMF和(c) Na@2HMF体系中初始*HMF氢化过程的动力学能垒。第一个HMF和第二个HMF代表HMF二聚体中的第一个和第二个HMF分子。(d) Na@2HMF体系中*HMFR生成*DHMF的基本步骤的自由能变化与电势的函数关系。(e) 在Li离子存在下,使用约束AIMD模拟沿反应坐标的自由能变化评估的水解离动力学能垒。(f) 在9 ps AIMD模拟过程中,2Li@2HMF体系中*H-Cu(111)上氢键长度的时间演化。
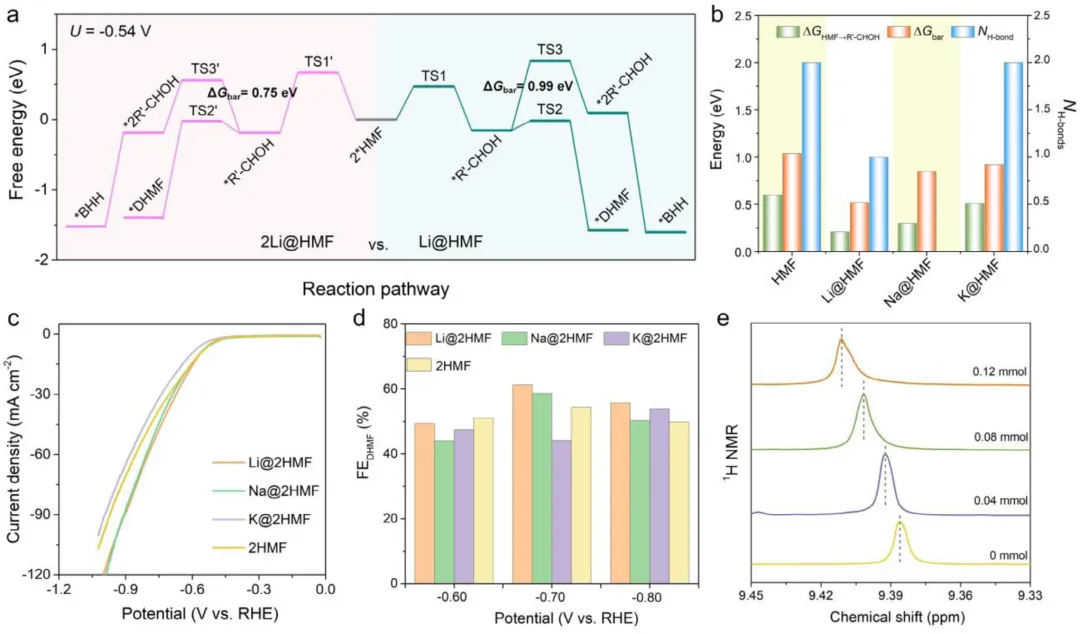
图5 (a) 在U = -0.54 V时,Li@2HMF和2Li@2HMF体系中HMFRR过程的自由能图。(b) 有和没有碱金属离子时*H-Cu(111)上HMFRR的理论自由能变化和动力学能垒:ΔG*HMF→*R-CHOH(*HMF氢化反应的自由能变化)、ΔGbar(在相应UL下的*HMF氢化反应动力学能垒)以及NH-bonds(两个HMF分子的氢键数目)。(c) 在含有0.15 M Li₂SO₄、Na₂SO₄和K₂SO₄的0.35 M (NH₄)₂SO₄电解质中,以及在不含碱金属的电解质(0.5 M (NH₄)₂SO₄,代表2HMF体系)中,Cu电催化剂在120 mM HMF下的LSV曲线和(d) 电势依赖的FE。(e) 在不同浓度Li⁺离子(0、0.04、0.08和0.12 mmol)下,HMF二聚体中醛基质子的¹H化学位移。
总之,通过最先进的恒电势DFT计算和AIMD模拟,该研究建立了一个统一的机理框架,用于理解高浓度条件下HMF在Cu催化剂上的还原过程。研究揭示,在工作电势下,HMF分子间的分子间氢键有效屏蔽了醛基,导致在高HMF浓度下HMFRR的动力学受到抑制。这一机理认识使得能够通过可控引入碱金属阳离子来合理调控界面微环境。在所研究的体系中,Li@2HMF表现出最高HMFRR活性,这是由于Li⁺能有效破坏HMF二聚体中的分子间氢键。这一效应得益于Li⁺的小离子半径、其深入内亥姆霍兹平面的能力、低配位结构以及其强吸电子能力,这些共同促进了H转移,同时在中等Li⁺浓度下保持了高HMF选择性。值得注意的是,电化学测量和¹H NMR光谱与理论预测完全一致,增强了所提出机理的稳健性。该研究阐明了分子间氢键如何控制电催化反应活性,并强调了碱金属阳离子在高反应物浓度下促进ECH的关键作用。这些发现预计将指导开发更高效的催化策略,用于生物质衍生平台分子的电化学升级。



 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
