



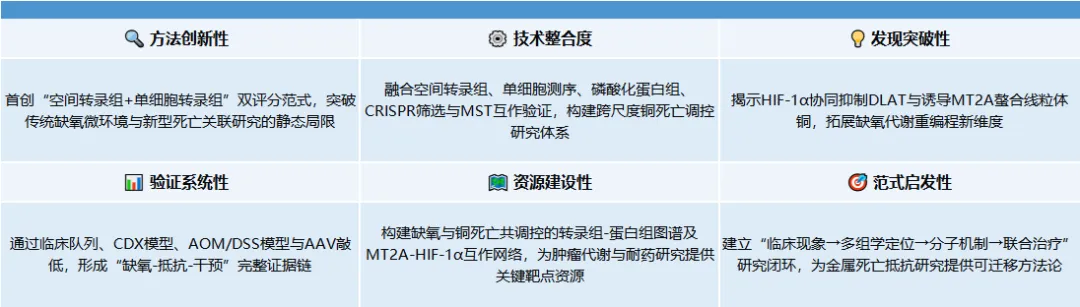

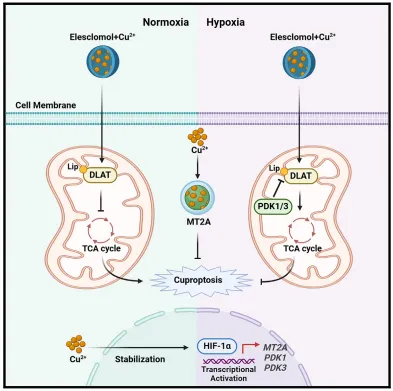
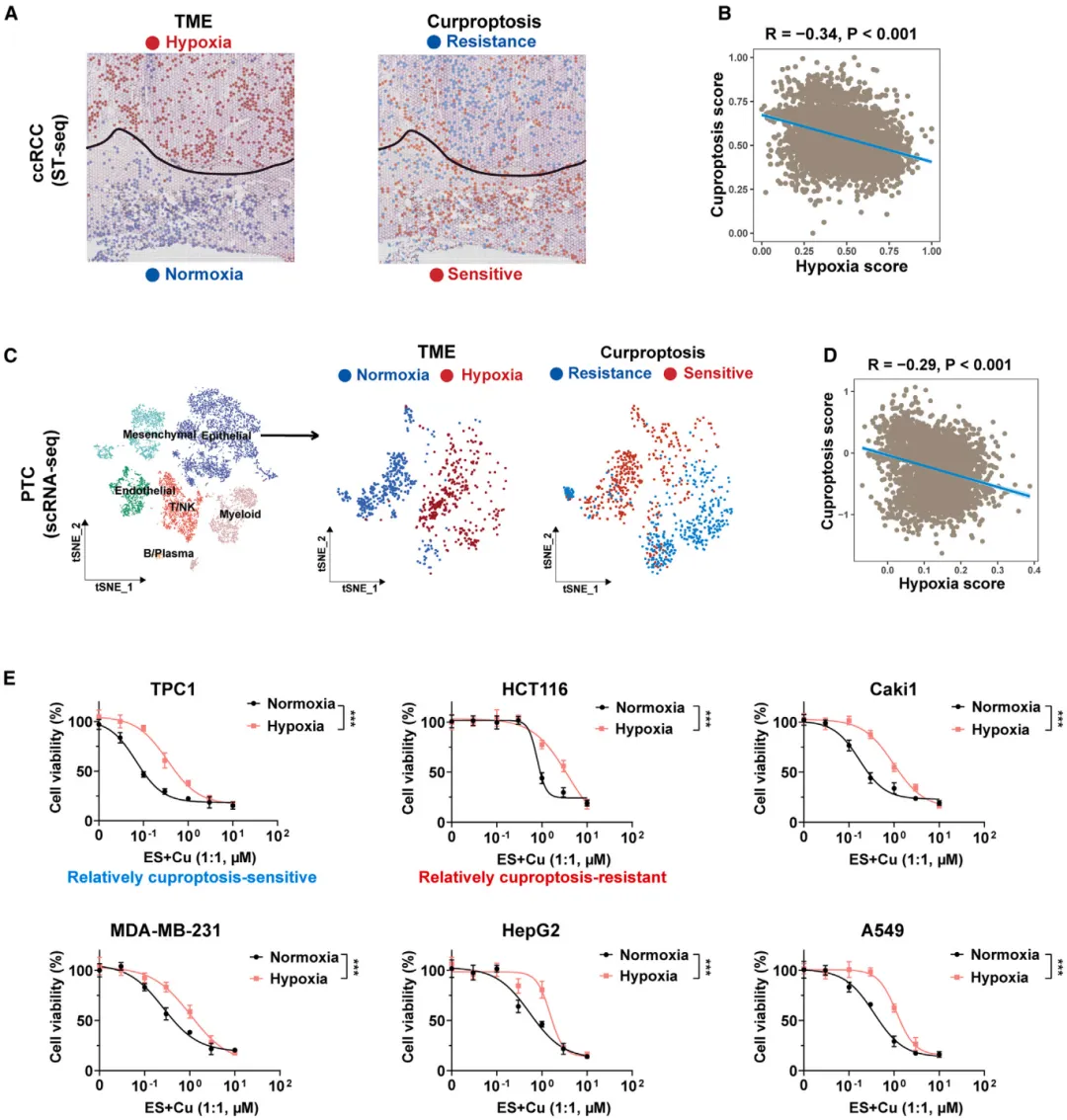
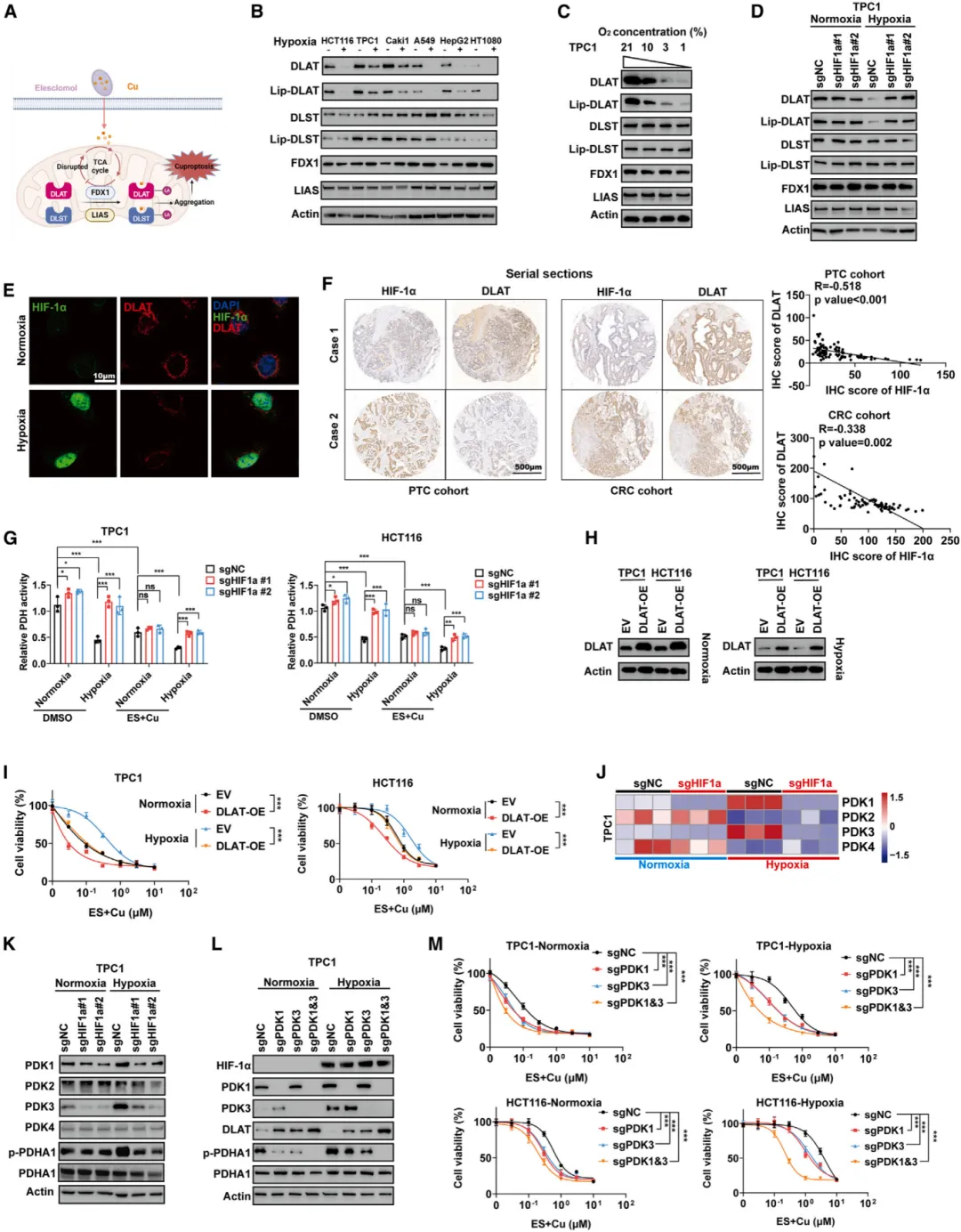




随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- 万象有声,触字生光—— 南京盲校学子亮相第五届视障读者 “光明阅读” 邀请赛大放异彩
- 一文说清南京小升初摇号,附2026小升初摇号难度预测!
- 南京富人区知名商场,即将拍卖
- 比赛通知 | 南京理工大学首届“兰杉杯”全英文模拟法庭辩论赛报名通知暨赛题发布
- 南京市栖霞区|丁家庄喜满满社区店
- 突发!落户要求变了!南京学区政策超重磅猛料传遍了!
- 南京湾顶豪实景现房横厅大平层六恒科技住宅
- 自己花心思整理的南京备忘录,都是自己或者身边朋友去过的挺不错的,在这分享给大家
- 可捡漏!南京三所“包分配”院校,分数不高,毕业端铁饭碗
- 江苏南京法院发布劳动典型案: 调岗 虚假报销 自认劳动关系 遭辱骂被迫离职 工伤 演出经纪关系
