作者
付更涛(南京师范大学)
引言
质子交换膜水电解(PEMWE)作为实现碳中和目标的关键技术,能够利用风能、太阳能等间歇性可再生能源高效生产绿色氢气。然而,阳极氧析出反应(OER)的缓慢动力学和酸性条件下的催化剂严重降解阻碍了PEMWE的大规模工业应用。尽管铱基氧化物(IrO2或IrOx)因其耐腐蚀性成为最先进的催化剂,但Ir的稀缺性和高成本以及其本征活性不足,严重制约了大规模部署。因此,开发低Ir载量、高活性和高稳定性的OER催化剂对于PEMWE至关重要。传统的吸附物演化机制(AEM)受限于OH*和OOH*吸附能之间的固有标度关系,理论最小过电位为0.37 V,限制了本征活性。因此,需要将催化机制转向绕过这些标度关系的新路径。在质子耦合电子转移(PCET)途径中,电子转移本质上与中间体的困难去质子化耦合并常受其限制。因此,加速去质子化过程是降低速率决定步骤(RDS)动力学障碍的基础,从而推动反应动力学超越热力学标度限制。桥氧(Obri)具有高电负性,使其成为热力学上更有利的质子受体,可以作为高效中继促进快速去质子化。然而,这一策略面临关键权衡:过高的酸性会导致从中间体中提取质子的驱动力不足,从而抑制去质子化动力学。因此,将Obri的质子亲和性优化到适中水平对于平衡质子提取和释放过程至关重要。双钙钛矿(A2B'B''O6)因其独特的结构灵活性和催化潜力而备受关注。其中,Ba2LnIrO6作为解决酸性OER机理挑战的理想模型系统脱颖而出。一方面,三价态(Ln3+)的引入通过电荷补偿机制促进活性高价铱(Ir5+)物种的形成,同时增强Obri的电负性以促进质子捕获。另一方面,Ln由于其高度局域化的4f轨道和5s、5p电子的屏蔽效应而表现出独特的物理性质,允许与配位环境杂化。在稀土-氧-过渡金属(RE-O-TM)单元中,特定的4f-2p-3d梯度轨道耦合有效地促进电荷转移,并通过调节活性位点的d带中心优化中间体的结合强度。理论上,通过Ln引入进行的这种电子调节预计将显著降低OOH*中间体的形成能。此外,Ba2LnIrO6双钙钛矿的独特结构创造了坚固的三维角共享八面体网络,可以增强热力学稳定性。然而,对于Ln的具体性质如何调节质子受体Obri位点与活性Ir中心的吸附行为之间的协同相互作用,仍缺乏全面的理解。
核心发现
本研究开发了一种有序Ba2EuIrO6双钙钛矿作为有效的OER催化剂,提出Ir-Obri-Eu单元作为关键活性中心,其中Obri作为质子受体发挥作用。获得的Ba2EuIrO6在10 mA cm-2下实现了250 mV的低过电位和1.39 A mg-1的高质量活性,优于BaIrO3和商业IrO2。原位光谱技术、同位素标记测量和自旋极化DFT计算证明了Ir-Obri-Eu单元作为活性中心的关键作用。Eu诱导的电子积累赋予Obri位点强质子亲和性,有利于从OER中的OOH*和OH*中间体捕获质子,触发桥氧介导的去质子化(BOMD)机制。此外,Eu的引入调节Ir dz2轨道以增加吸附氧(Oads)的自旋密度,加速-OH攻击并降低OOH*形成的能垒。组装的Ba2EuIrO6 PEMWE在1.67 V下达到1.0 A cm-2,并在1.0 A cm-2下稳定运行350小时,展示了其实际应用潜力。
图文解读
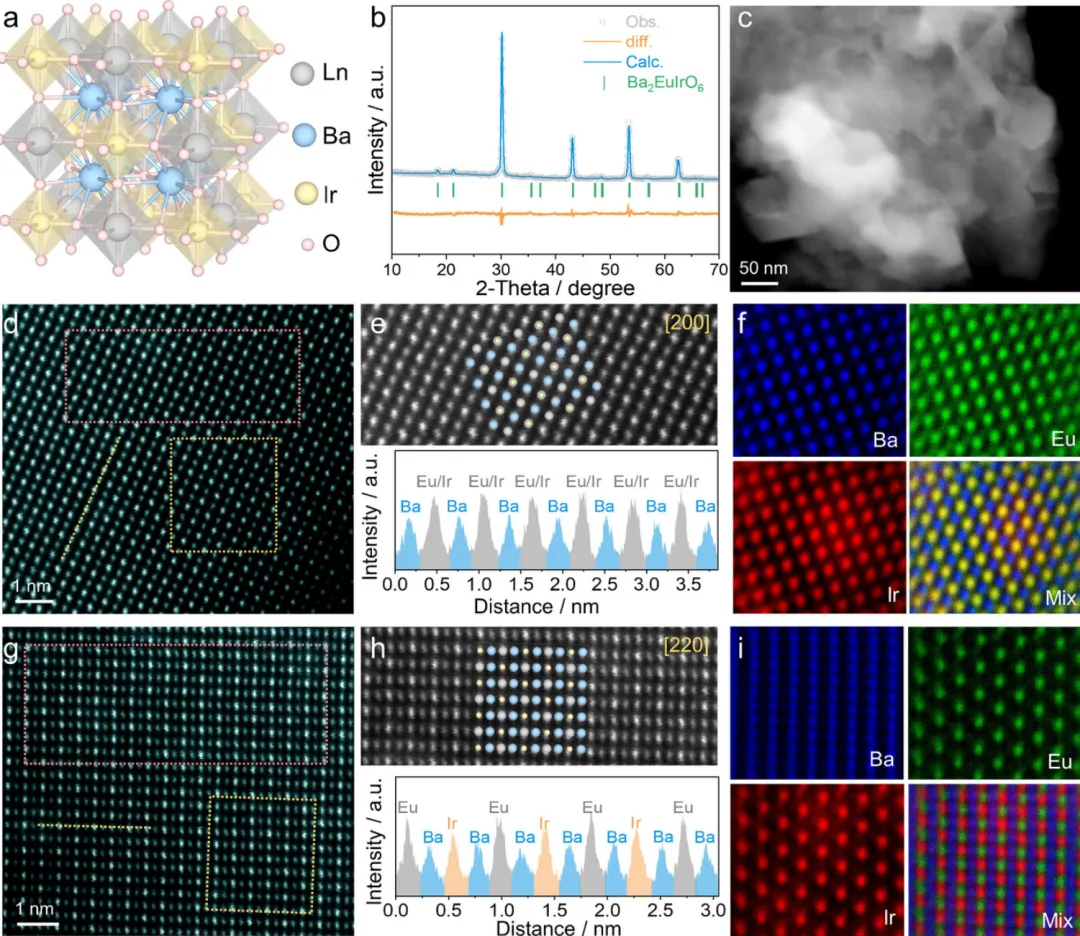
图1:Ba2EuIrO6双钙钛矿的晶体结构和原子排列表征
图1展示了Ba2EuIrO6双钙钛矿的晶体结构和原子排列特征。图1a展示了通用Ba2LnIrO6的晶体结构模型,显示了双钙钛矿的典型特征。图1b通过Rietveld精修XRD图谱确认了制备的Ba2EuIrO6具有典型的立方结构。高角环形暗场扫描透射电子显微镜(HAADF-STEM)分析表明,制备的Ba2EuIrO6由纳米颗粒聚集体组成(图1c)。球差校正HAADF-STEM(AC-HAADF-STEM)图像展示了Ba2EuIrO6中暗Ba原子和亮Ir/Eu原子的有序堆叠序列(图1d)。沿[200]晶带轴观察的实验原子排列与相应的模拟原子模型一致(图1e)。亮的混合原子柱由交替的Eu和Ir原子组成,位于由四个较暗Ba柱形成的方形排列的中心。从图1d中黄线提取的线强度分布说明了Ba和Ir/Eu原子的有序排列。原子分辨率能量色散X射线光谱(EDS)元素映射进一步确认了Ba和Ir/Eu原子的有序排列,清楚地显示了Eu和Ir原子在同一柱内的空间叠加(图1f)。Ba2EuIrO6的晶体结构和原子排列也在[220]晶带轴中揭示(图1g),与模拟的原子排列一致(图1h)。Ba原子在明显的柱中排列,而Ir和Eu原子在相邻柱内交替排列。原子分辨率EDS元素映射图像(图1i和S3)确认了沿[220]晶带轴的Ba、Eu和Ir原子的有序排列。这些结果确认了获得的Ba2EuIrO6的高度有序双钙钛矿结构。作为对比,通过相同工艺合成了BaIrO3,只是省略了Eu前驱体。Rietveld精修XRD图谱和结构表征确认了BaIrO3相的成功合成(图S4和S5)。这些表征结果充分证明了Ba2EuIrO6的成功制备和其高度有序的双钙钛矿结构,为后续的催化性能研究奠定了基础。
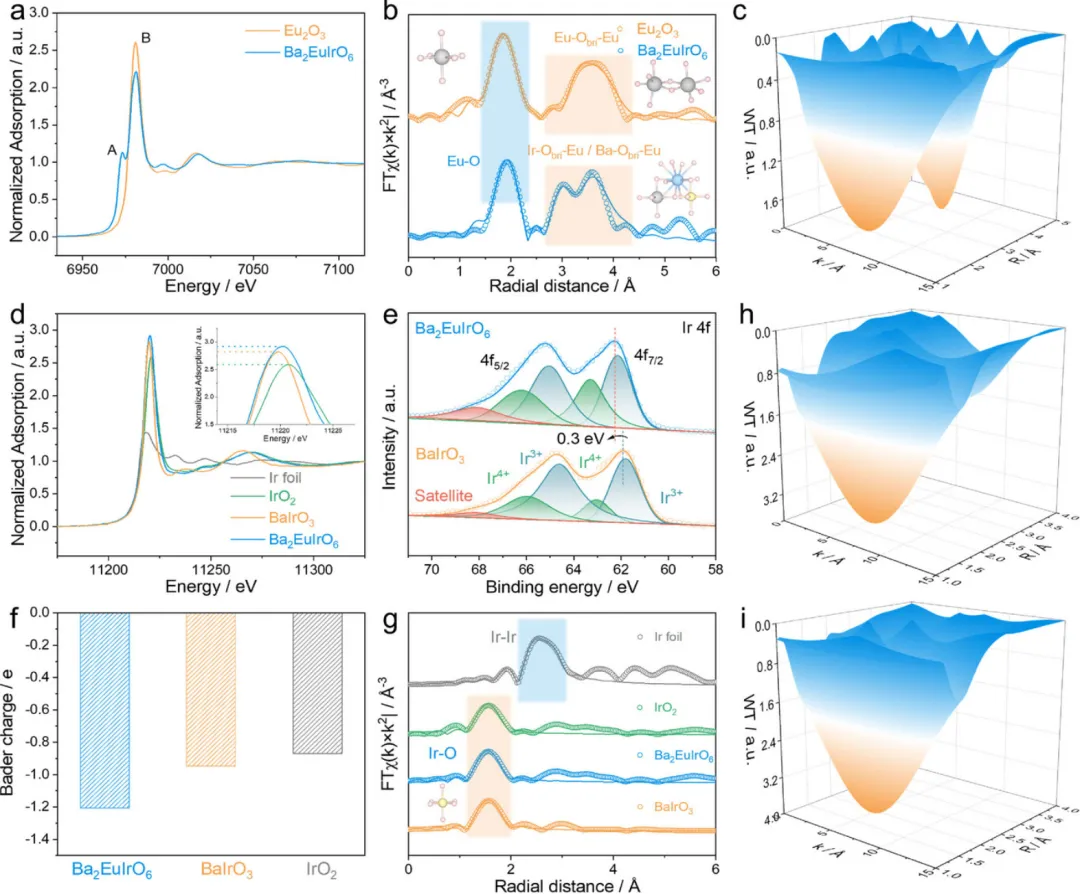
图2:Ba2EuIrO6的电子结构和化学状态分析
图2通过X射线光电子能谱(XPS)和X射线吸收谱(XAS)深入分析了Ba2EuIrO6的表面化学和电子状态。图2a展示了Ba2EuIrO6和Eu2O3的归一化Eu L3边X射线吸收近边结构(XANES)谱。Eu2O3样品表现出特征峰(峰B),归因于从2p3/2到5d的电子跃迁,与Eu3+(4f6)相关。与Eu2O3相比,Ba2EuIrO6的白线强度较低,并且出现了归属于Eu2+的明显峰(峰A),确认Ba2EuIrO6中的Eu具有比Eu2O3更低的氧化态Euδ+(2<δ<3)。傅里叶变换扩展X射线吸收精细结构(FT-EXAFS)进一步分析了Eu的局部原子环境。如图2b所示,与Eu2O3(1.84 Å)相比,Ba2EuIrO6显示出增加的Eu-O键长(1.92 Å),这是由于EuO6多面体的特定结构约束和对称性演变。Ba2EuIrO6的第二壳层峰涉及Ir-Obri-Eu和Ba-Obri-Eu相互作用,与Eu2O3中的Eu-Obri-Eu环境不同,这与小波变换(WT)-EXAFS谱一致(图2c)。图2d展示了Ba2EuIrO6、BaIrO3和选定参考样品的Ir L3边XANES谱。Ba2EuIrO6的白线强度比BaIrO3和IrO2更高,表明Ir氧化态更高。这一现象归因于Eu3+的引入,驱动Ir采用更高的价态以保持晶格电中性。该结果与Ir 4f XPS谱一致(图2e),其中Ba2EuIrO6的Ir 4f7/2峰相对于BaIrO3向更高结合能位置移动了0.30 eV。高价Ir5+的稳定需要氧配体上的高电子密度,从而增强Obri的电负性。Bader电荷分析为这种电子重分布提供了有力证据(图2f)。Ba2EuIrO6中Obri的计算电荷为-1.21 |e|,比BaIrO3和IrO2更负,确认了负电荷的积累,预计这将调节关键OER中间体的吸附行为,从而可能优化反应动力学。此外,Ba2EuIrO6和BaIrO3的WT-EXAFS和FT-EXAFS谱揭示了明显的第一壳层Ir-O配位,几乎没有第二壳层特征,与钙钛矿晶格内几何孤立的IrO6单元的存在一致(图2g-i)。这些结果充分证明了Eu的引入成功调节了Ir的电子结构和Obri的电负性,为后续的高效OER催化性能奠定了电子结构基础。
图3:Ba2EuIrO6的电催化OER性能评估
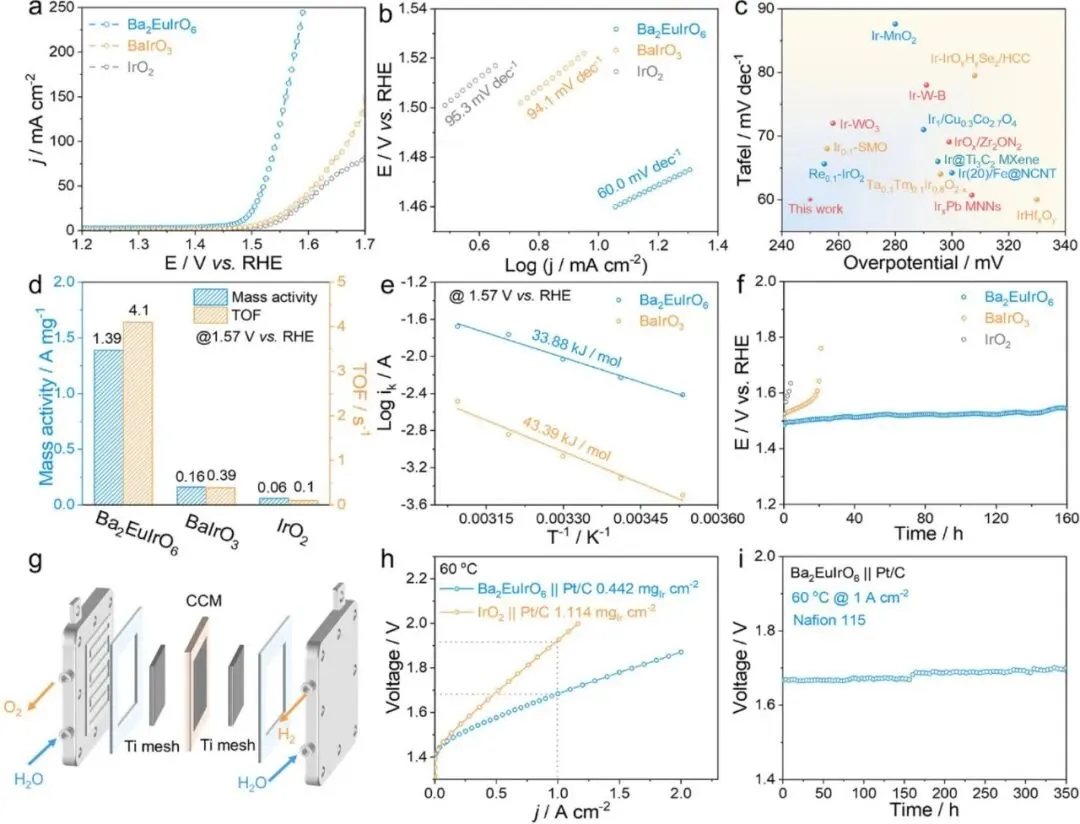
图3全面评估了Ba2EuIrO6在酸性条件下的电催化OER性能,并与BaIrO3和商业IrO2进行了对比。图3a展示了催化剂在O2饱和的0.5 M H2SO4电解液中的线性扫描伏安法(LSV)曲线,其中Ba2EuIrO6表现出最高的电流响应。Ba2EuIrO6在10 mA cm-2下的过电位仅为250 mV,优于BaIrO3(300 mV)和IrO2(310 mV)。通过Tafel斜率分析评估了催化剂的氧析出动力学(图3b)。Ba2EuIrO6显示出60 mV dec-1的较低Tafel斜率,低于BaIrO3(94.1 mV dec-1)和IrO2(95.3 mV dec-1),表明更有利的反应动力学。Ba2EuIrO6的OER活性也优于一系列先前报道的Ir基OER催化剂(图3c和表S1)。为研究本征电催化活性,计算了质量活性和转换频率(TOF)。如图3d所示,Ba2EuIrO6在1.57 V vs. RHE下的质量活性为1.39 A mg-1,分别是BaIrO3和IrO2的8.7倍和23.2倍。基于Ir作为关键活性位点计算的Ba2EuIrO6的TOF值为4.1 s-1,优于对照样品。采用电化学双层电容(Cdl)评估电化学活性表面积(图S7)。Ba2EuIrO6表现出比BaIrO3(6.0 mF cm-2)高得多的Cdl值(24.5 mF cm-2),表明Ba2EuIrO6的比活性高于BaIrO3。从基于温度依赖OER活性的Arrhenius型图推导出催化剂的表观活化能(Eapp)(图S8)。如图3e所示,Ba2EuIrO6的Eapp值计算为33.88 kJ mol-1,低于BaIrO3(43.39 kJ mol-1),表明Ir-Obri-Eu单元降低了OER过程的能垒。除了催化活性外,稳定性对于评估OER催化剂的实际应用至关重要。通过在OER电位窗口内连续循环伏安法(CV)扫描进行加速耐久性测试(ADT)。Ba2EuIrO6的LSV在3000次循环测试后呈现可忽略的电流衰减,而BaIrO3表现出明显的活性下降(图S9)。此外,计时电位法(CP)测试也验证了Ba2EuIrO6的更好稳定性,在160小时后电位略有增加(图3f)。i-t测试后,XRD结果表明回收的Ba2EuIrO6的双钙钛矿相结构没有发生任何显著变化(图S10a),表明Ba2EuIrO6中角共享IrO6八面体的三维网络的坚固性。回收的BaIrO3中Ir 4f的XPS结合能相对于初始样品显示出0.60 eV的正移,而Ba2EuIrO6仅显示出0.22 eV的微小正移,表明Eu有利于稳定Ir的高氧化态(图S10b,c)。受Ba2EuIrO6高OER性能的鼓舞,组装了以制备的催化剂为阳极、40 wt% Pt/C为阴极的PEMWE以评估工业应用潜力(图3g)。具体而言,Ba2EuIrO6 || Pt/C仅需1.68 V的电池电压即可达到1 A cm-2的电流密度,明显低于IrO2 || Pt/C(1.91 V)(图3h)。PEMWE中Ba2EuIrO6阳极的Ir质量负载为0.442 mgIr cm-2,低于IrO2阳极(1.114 mgIr cm-2),展示了低Ir载量设计。此外,使用Ba2EuIrO6 || Pt/C的PEMWE电池可以在60°C下以1 A cm-2运行350小时,电压略有增加,呈现出良好的长期稳定性(图3i)。PEMWE中的计算能耗为44.7 kWh kg-1 H2,突显了基于Ba2EuIrO6的PEMWE的高能效。这些性能数据充分证明了Ba2EuIrO6作为高效OER催化剂的巨大潜力。
图4:Ba2EuIrO6的OER机理原位电化学研究
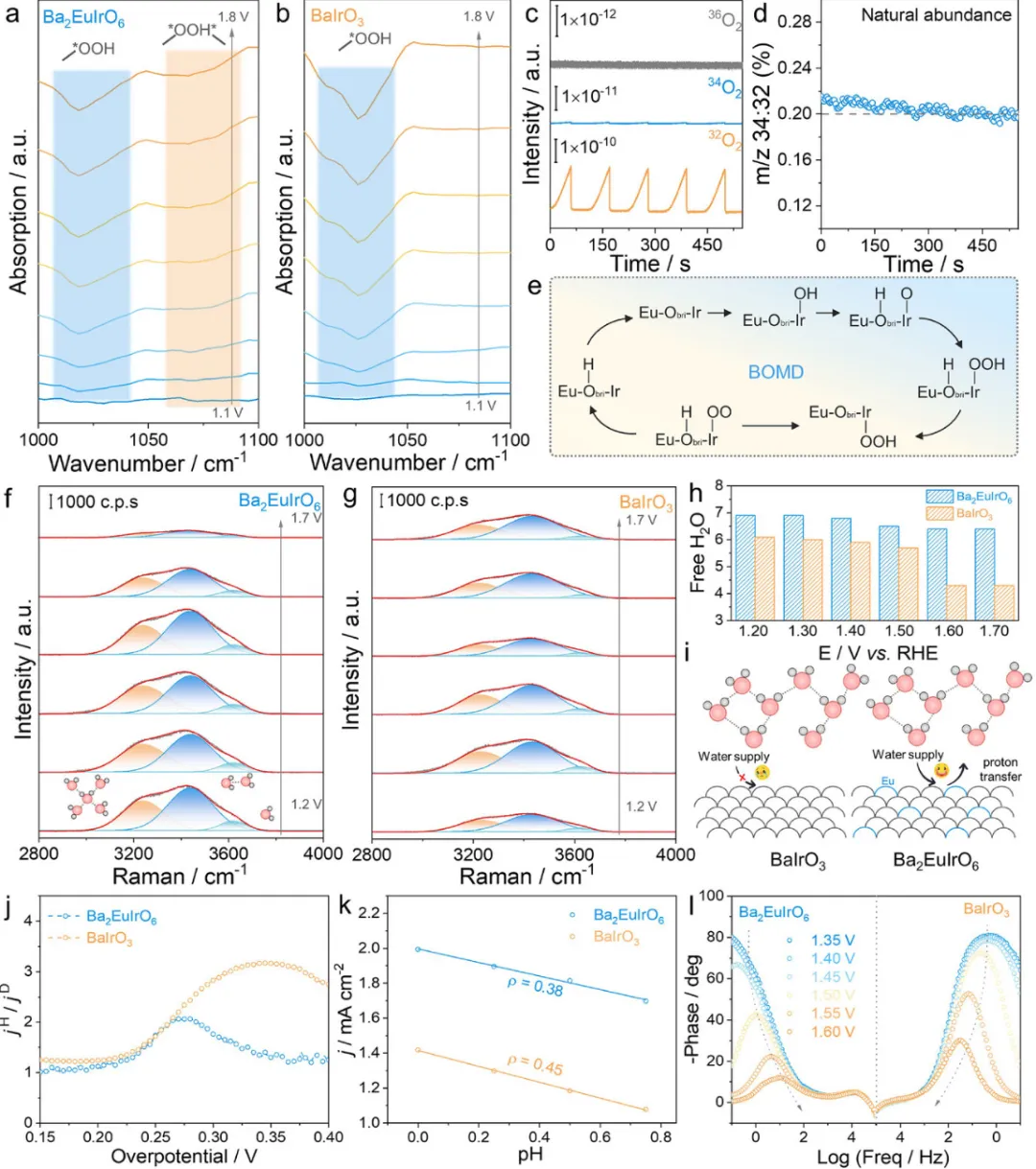
图4通过多种原位电化学技术深入研究了Ba2EuIrO6的OER机理。图4a,b展示了原位衰减全反射表面增强红外吸收光谱(ATR-SEIRAS)结果。当达到预OER区域(1.2 V)时,Ba2EuIrO6和BaIrO3在约1020 cm-1处表现出宽峰,归因于酸性OER期间关键中间体OOH*的形成,确认了遵循动力学缓慢的AEM途径。与BaIrO3相比,Ba2EuIrO6在1060 cm-1处出现了一个新的肩峰,归因于OOH*与邻近分子之间氢键的分子振动,证明了额外的去质子化步骤(OOH*→OO*-H*)。如前所述,Obri具有强电负性并作为质子受体从OOH*中间体捕获H*,这与原位ATR-SEIRAS一致。为阐明Ba2EuIrO6的OER反应途径,进行了耦合同位素标记的操作差分电化学质谱(DEMS)测量以分析气体产物。最初,在H218O电解液(0.5 M H2SO4)中在1.2-1.6 V电压范围内通过CV用18O标记Ba2EuIrO6。随后,用去离子水彻底冲洗18O标记的催化剂,并在H216O电解液中进行多次LSV扫描。在此过程中,监测了32O2、34O2和36O2的信号。如图4c所示,32O2是主要产物,未检测到36O2。此外,34O2/32O2的比值与自然丰度一致(约0.2%,图4d),确认生成的氧气来源于水而非晶格氧。据此,提出了Ir-Obri-Eu单元处的桥氧介导去质子化(BOMD)途径,如图4e所示。最初,Ir位点作为活性中心被OH*攻击,然后Obri帮助质子转移形成O*。接下来,OH*吸附在O*上形成OOH*。随后,OOH*的质子转移到O2*,随后O2最终脱附。在该机制中,Obri作为质子受体,规避了与单一活性位点相关的传统标度关系,从而增强OER活性。在水溶液中,质子转移主要通过Grotthuss机制进行,其中质子沿氢键传递而没有水分子本身的显著物理位移,表现出比载体扩散更高的效率。因此,界面氢键网络的连通性显著影响质子转移速率。进行了原位拉曼光谱技术以定量研究界面水的结构演变(图4f,g)。拉曼峰可以分解为三种类型的振动模式,位于约3200、约3400和约3600 cm-1,分别归属于四氢键水(4-HB·H2O)、二氢键水(2-HB·H2O)和自由H2O。在预催化阶段,与BaIrO3相比,Ba2EuIrO6观察到更强的界面水信号,证明由于Eu的高亲氧性而富集吸附的H2O。随着电位增加,Ba2EuIrO6上的界面水信号迅速降低,进一步证实Ir-Obri-Eu单元加速OER动力学。此外,Ba2EuIrO6中自由水的比例保持相对稳定,而BaIrO3中的比例显示出明显下降。该结果表明Eu在调节界面水中发挥双重作用。一方面,Eu的高亲氧性增强水吸附并富集活性Ir-Obri-Eu单元周围的H2O分子。另一方面,Eu重塑局部氢键环境,从而在OER期间保持相对稳定的自由H2O比例,确保在界面连续供应易反应的水物种(图4h)。同时,由Ir-Obri-Eu单元诱导的重构氢键网络变得更加有序和动态互连,有利于质子跨界面水层传递,从而促进OER期间的质子转移动力学(图4i)。为深入了解酸性OER中增强的质子转移特性,进行了氘同位素标记实验、pH依赖性测量和原位电化学阻抗谱(EIS)测量。通过比较0.5 M H2SO4(H2O)和0.5 M D2SO4(D2O)中的OER活性评估动力学同位素效应(KIE)。用氘取代质子会影响质子转移过程的动力学,如图S11所示。可以基于H2O与D2O的OER电流密度比计算KIE值。与BaIrO3相比,Ba2EuIrO6的KIE值较小(图4j),表明Obri在加速去质子化过程中的有益作用。图S12显示了Ba2EuIrO6和BaIrO3在不同pH下的LSV测量。根据特定电压(1.57 V vs. RHE)下电流密度的偏导数计算pH的反应级数(ρ)。显然,BaIrO3的ρ(0.45)高于Ba2EuIrO6(0.38),表明OER过程中更高效的PCET过程(图4k)。此外,进行了原位EIS测量以研究界面反应动力学。如Ba2EuIrO6的Bode图所示(图4l),低频区的小相角对应更快的OER动力学。随着电位增加,Ba2EuIrO6的相角显著降低,而BaIrO3显示出更慢的下降。这些结果确认了伴随高效表面去质子化耦合电子转移过程的快速OER动力学。
图5:Ba2EuIrO6的OER机理理论分析
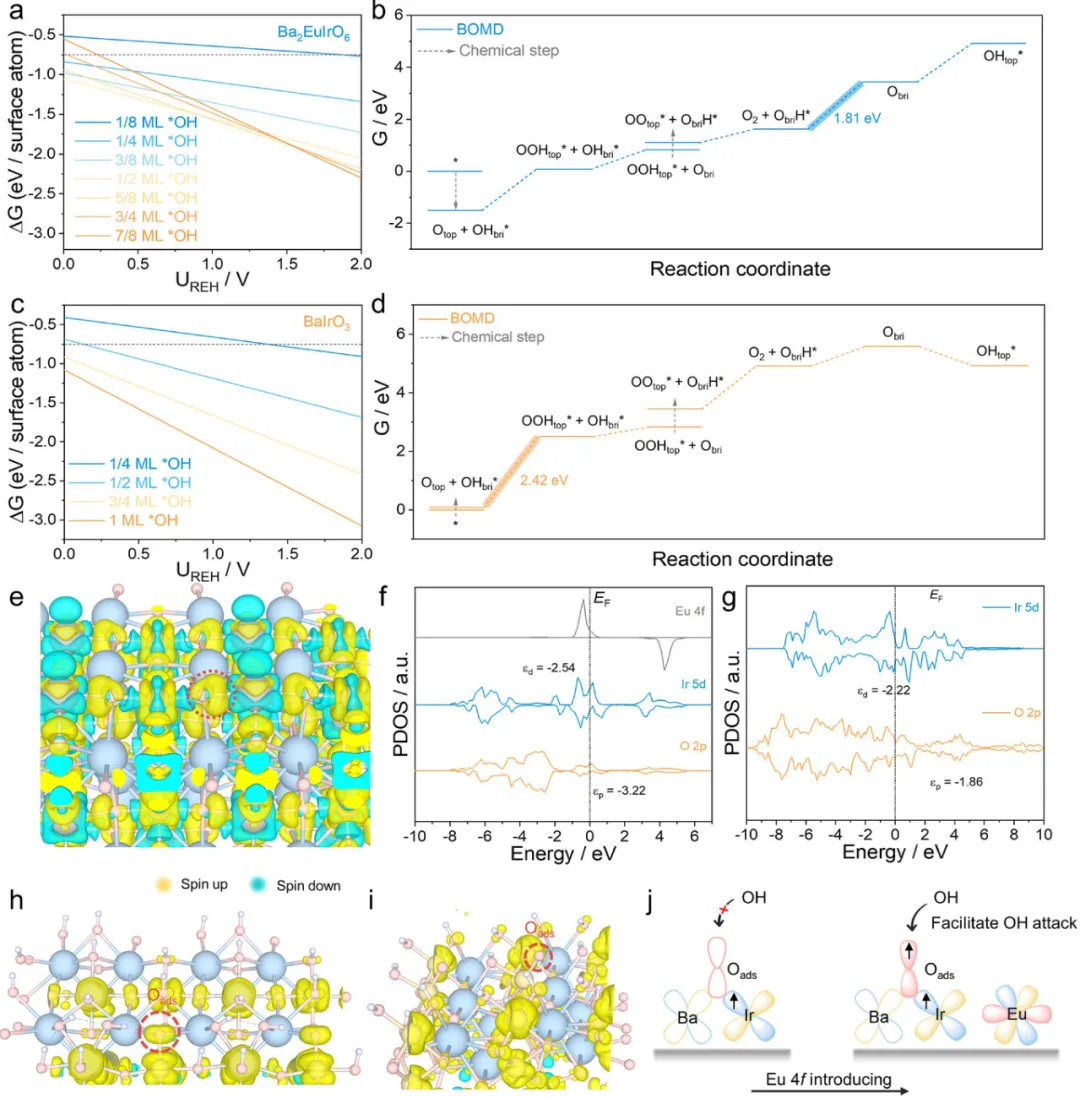
图5通过自旋极化密度泛函理论(DFT)计算深入分析了Ir-Obri-Eu对OER活性的影响。考虑到电化学反应条件下表面状态的变化,计算了OH*物种的预覆盖以模拟更真实的反应相。在1/8到7/8 ML OH*的覆盖范围内计算筛选了所有潜在的OH*吸附位点(图S13)。根据1D Pourbaix图(图5a),7/8 ML OH*是实验OER电位窗口内最稳定的OH*覆盖度。为从理论上阐明OER机理,系统研究了LOM、AEM和BOMD途径(图S14)。对于Ba2EuIrO6,由于氧空位(Ov)形成的潜在决定步骤(PDS)为3.06 eV,LOM途径在热力学上不利。对于AEM途径,从OOH*演化O2作为PDS,能垒较低为2.43 eV。鉴于Obri在中间体去质子化中的关键作用,在BOMD途径中,Ir-OHads*的质子化学转移到邻近的Obri位点(图5b和S15),呈现-1.50 eV的放热变化,确认Obri的高质子亲和性。下一步是OOH*的形成和H*转移到Obri,自由能变化为0.52 eV。与AEM相比,PDS计算为ObriH*的去质子化步骤,自由能变化为1.81 eV,低于AEM中的PDS 2.43 eV,证明BOMD机制显著优于LOM和AEM。为进一步验证Ir-Obri-Eu单元对OER动力学的促进机理,研究了BaIrO3和IrO2上的反应机理。图5c展示了BaIrO3的OH*覆盖Pourbaix图,其中1 ML OH*是最稳定的相。随后,评估了LOM和AEM途径(图S16)。与Ba2EuIrO6类似,Ov的生成和O2的脱附作为LOM和AEM的PDS,能垒分别为4.23和2.66 eV。对于BOMD途径,OOH*的形成构成PDS,能垒为2.42 eV(图5d)。因此,可以得出结论,具有特定Ir-Obri-Eu局部环境的OER有利于BOMD途径。与先前报道一致,IrO2遵循AEM途径。考虑OH*覆盖后,O2脱附仍然是PDS,能垒为2.47 eV(图S17和S18)。也尝试模拟IrO2的BOMD机制。然而,在ObriH-Ir-OO*中间体的结构优化过程中,Obri上的H*不稳定并自发转移回OO*以重新形成OOH*(图S19)。Bader电荷分析(图2f)揭示IrO2中的Obri具有最低的电子密度,使得Ir-Obri-Ir无法诱导BOMD所需的质子迁移,确认Obri的适当质子亲和性是BOMD途径的前提。进一步探讨了Eu引入对本征电子结构的影响。Ba2EuIrO6的电荷密度差(图5e)揭示了Obri上明显的电子积累,证实其高电子密度。此外,比较了Ba2EuIrO6和BaIrO3的投影态密度(PDOS)(图5f,g)。与BaIrO3相比,Ba2EuIrO6中Ir 5d和O 2p带的带宽变窄,表明Eu引入诱导Ir和O原子上增强的电子局域化,与电荷密度差结果完美一致。此外,Ba2EuIrO6的Ir d带中心(εd)仅显示出轻微的负移,而O p带中心(εp)显著下移,从-1.86到-3.22 eV,这可能是由费米能级(Ef)附近Eu 4f态的强占据驱动的(图S20)。较低的O 2p带中心意味着Obri上增强的电子局域化,促进质子亲和性。为进一步研究BaIrO3中OOH*形成的PDS起源,分析了自旋密度。如图5h,i所示,观察到Ba2EuIrO6的Oads上自旋密度显著增加,有利于接受-OH的亲核攻击并加速关键的OOH*形成步骤(图5j)。现代分子轨道成键理论表明,中间体的电子性质与dz2轨道的活性中心相关,这是由于σ型相互作用。如图S21所示,Ba2EuIrO6的Ir dz2轨道表现出比BaIrO3更高的电子占据,促进电子离域到Oads,从而产生具有明显自由基特征的氧物种。总之,理论研究确认了Ir-Obri-Eu单元的双重作用:Obri位点作为有效的质子受体触发BOMD途径,而调节的Ir dz2轨道电子结构增强Oads的自旋密度以降低OOH*的能垒。
总结
本研究报道了一种低Ir载量、高活性的Ba2EuIrO6催化剂,用于有效催化酸性电解液中的OER,并确定了Eu在促进催化转化中的关键作用。Ba2EuIrO6催化剂在10 mA cm-2下实现了250 mV的低过电位,并支持稳定的PEMWE运行。通过原位光谱研究、同位素标记测量和理论计算,发现Ir-Obri-Eu单元作为催化活性中心发挥关键作用。Ir-Obri-Eu单元具有强质子亲和性,用于从OOH*和OH*捕获质子,触发桥氧介导的去质子化机制,打破OER期间的传统标度关系。此外,Eu的引入调节Ir dz2轨道以增加吸附氧的自旋密度,加速-OH攻击并降低OOH*形成的能垒。Ba2EuIrO6负载的PEMWE在仅1.67 V下提供超过1.0 A cm-2,并在1.0 A cm-2下稳定运行350小时,展示了其实际应用的良好潜力。
原文链接
https://doi.org/doi.org/10.1002/anie.8724105