心肌梗死(心梗)后心脏自愈机制的颠覆性发现,并为未来挽救心衰提供了极其精准的“新药靶点”。 南京大学张龙江教授(东部战区总医院放射诊断科主任)与上海交通大学医学院附属仁济医院副院长卜军教授为共同通讯作者。

一、研究背景与目的
心肌梗死(MI)后巨噬细胞对凋亡心肌细胞的胞葬作用(efferocytosis) 是心脏修复、炎症消退的核心环节。CD40 为 TNF 受体超家族成员,胞内段有TRAF2/3/5与TRAF6两个结合位点,既往认为 CD40 主要介导炎症,但其在心梗后巨噬胞葬中的具体作用与下游通路尚不清晰。
本研究旨在明确:
二、实验设计与关键方法
实验模型 / 技术 | 核心用途 |
全身性 CD40 KO、髓系特异性 CD40 cko、巨噬细胞特异性 CD40 敲除小鼠 | 明确 CD40 的细胞特异性功能 |
CD40-TRAF2/3/5⁻/⁻、CD40-TRAF6⁻/⁻小鼠 | 区分两条下游通路的作用 |
单细胞 RNA 测序(scRNA-seq) | 分析巨噬亚群、修复前体细胞 |
超声、9.4T 心脏 MRI、TTC/Masson 染色 | 评估心功能、心梗面积、纤维化 |
流式、免疫荧光、WB、ELISA | 检测胞葬、炎症、蛋白表达 |
腺病毒转染 ΔCD40(保留 TRAF2/3/5、缺失 TRAF6) | 验证靶向通路的治疗效果 |
三、核心表型发现:CD40是心梗后修复的“保护因子”
(一)CD40 在心梗后心脏的表达特征
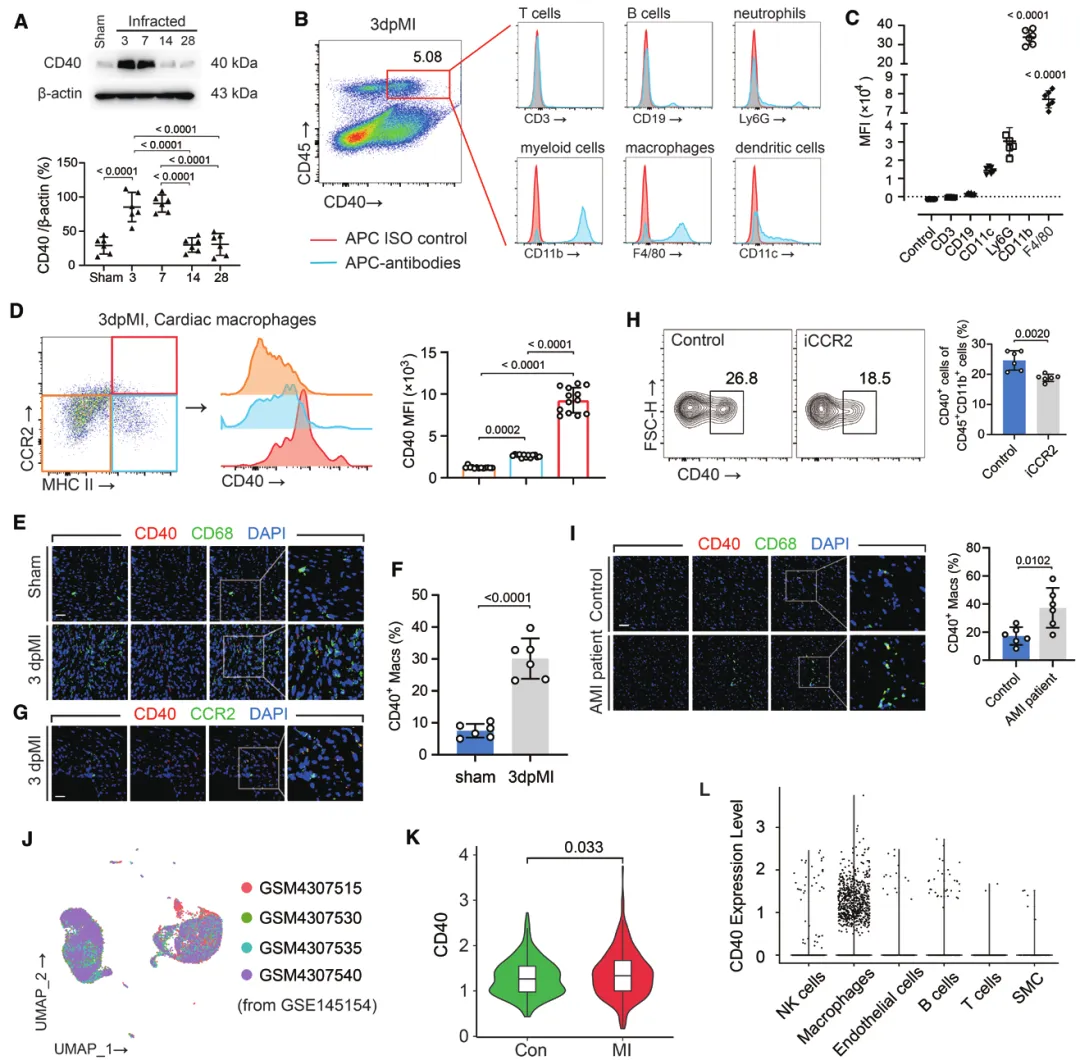
(二)CD40 缺失加重心功能损伤
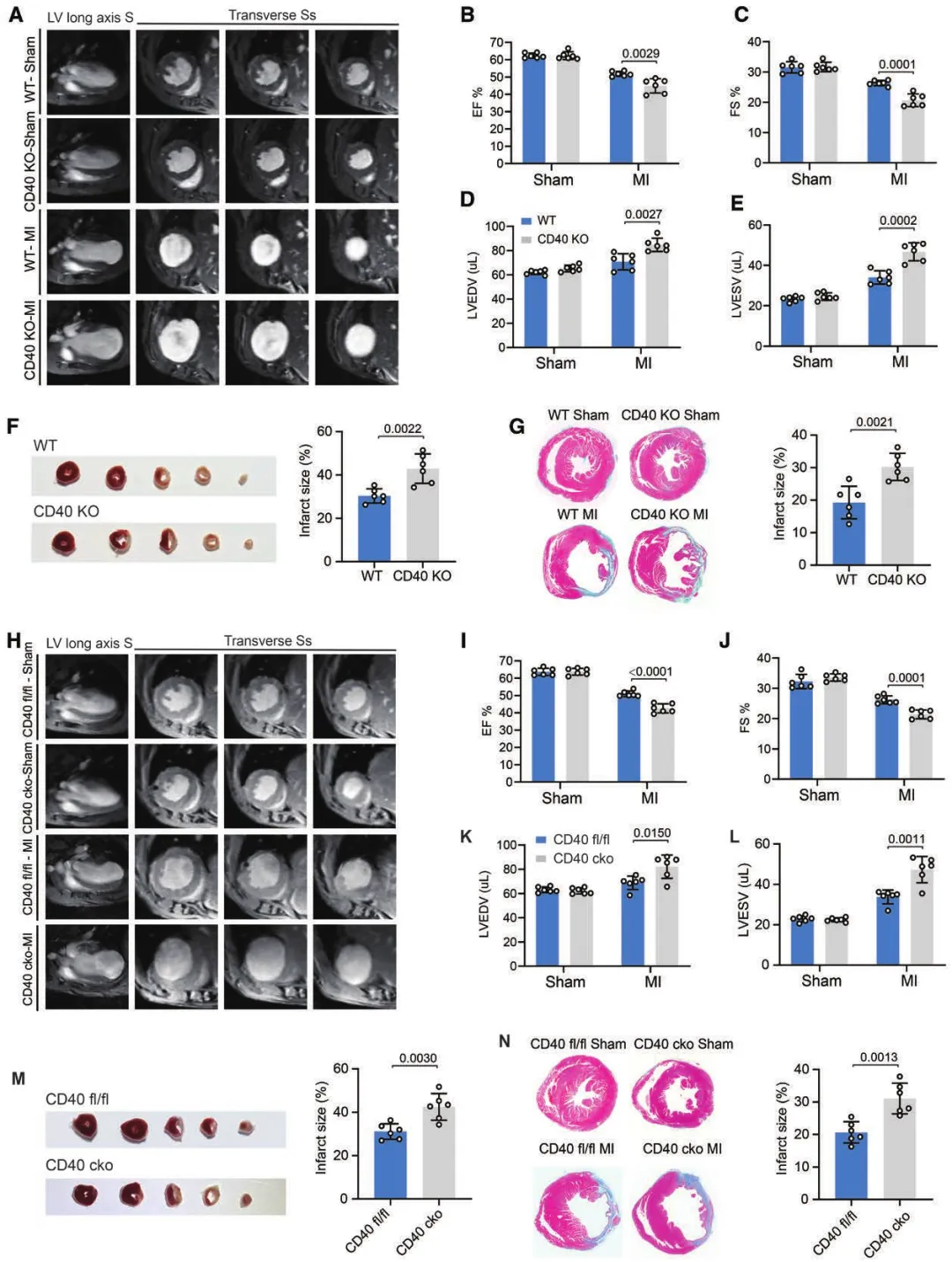
全身性 / 髓系 / 巨噬细胞特异性 CD40 敲除小鼠验证:所有CD40缺失小鼠心梗后梗死面积更大、左心室扩张更明显、射血分数显著降低;缺血再灌注(IR)模型中,髓系CD40缺失同样加剧心功能恶化;sham手术组无差异,说明CD40的功能仅在损伤状态下发挥。
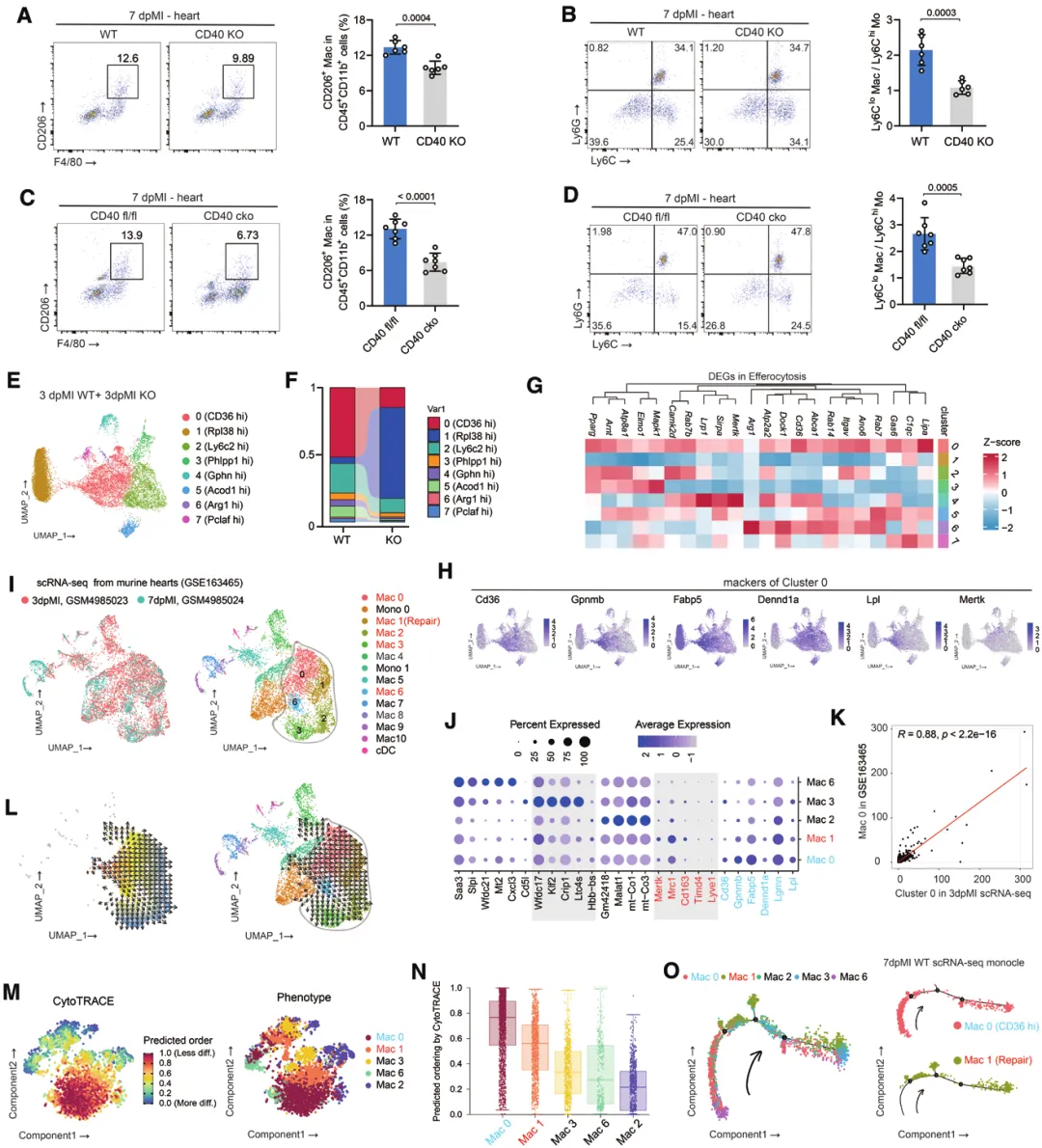
四、机制解析:CD40通过TRAF2/3/5→STAT6轴促进胞葬
(一)CD40缺失损害巨噬细胞胞葬与修复表型转化
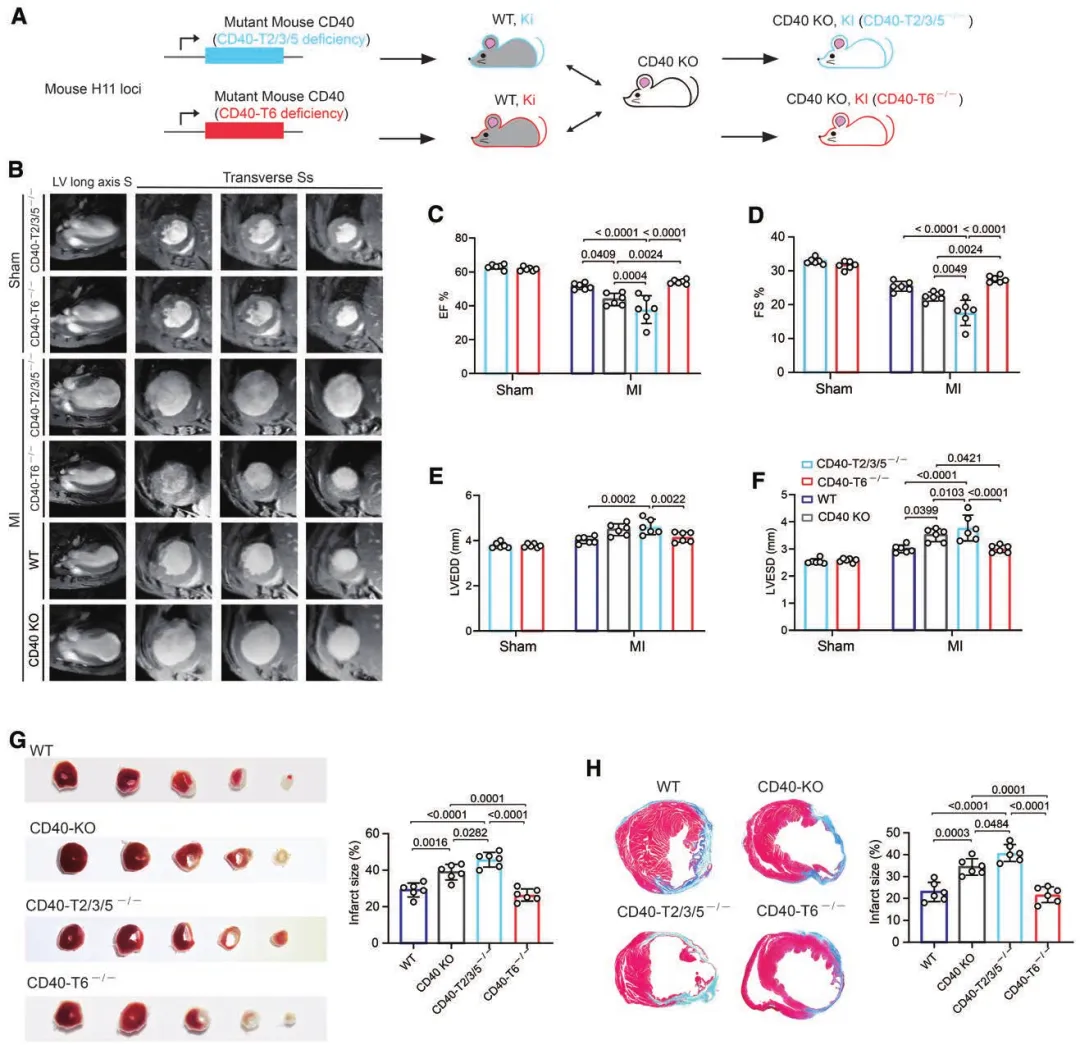
(二)两个TRAF位点的功能分离
直接激活CD40(使用激动性抗体FGK4.5)无法增强胞葬,反而诱导强烈促炎表型,证明单纯激活CD40整体无修复获益;
构建特异性突变小鼠:仅保留TRAF2/3/5位点的CD40可完全挽救CD40缺失的心功能损伤,仅保留TRAF6位点的CD40则无保护作用。
(三)下游机制:STAT6是核心转录因子
蛋白组学+生信分析发现:CD40-TRAF2/3/5信号激活STAT6,磷酸化STAT6入核,直接结合MerTk和Arg1的启动子区域,上调二者表达,最终增强巨噬细胞胞葬能力;使用STAT6抑制剂可完全消除TRAF2/3/5对胞葬的促进作用。
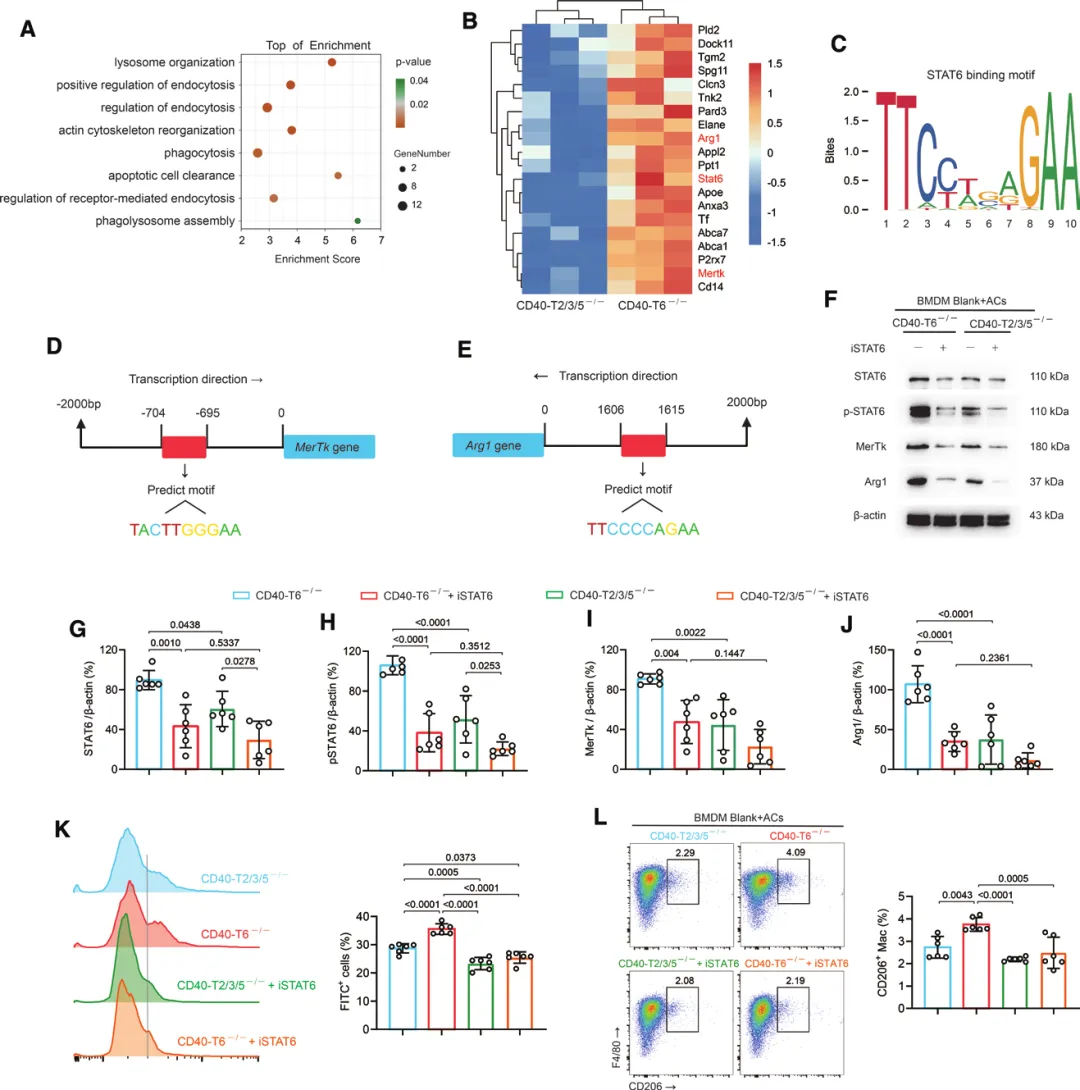
(四)治疗验证:精准激活TRAF2/3/5通路有效
用携带F4/80启动子的腺病毒,在CD40 cko小鼠的巨噬细胞中特异性过表达仅保留TRAF2/3/5结合位点的CD40突变体(ΔCD40):显著减少梗死面积、改善左心室功能;提升巨噬细胞胞葬效率、增加修复型巨噬细胞比例、促进新生血管形成和早期纤维化修复、降低全身炎症水平。
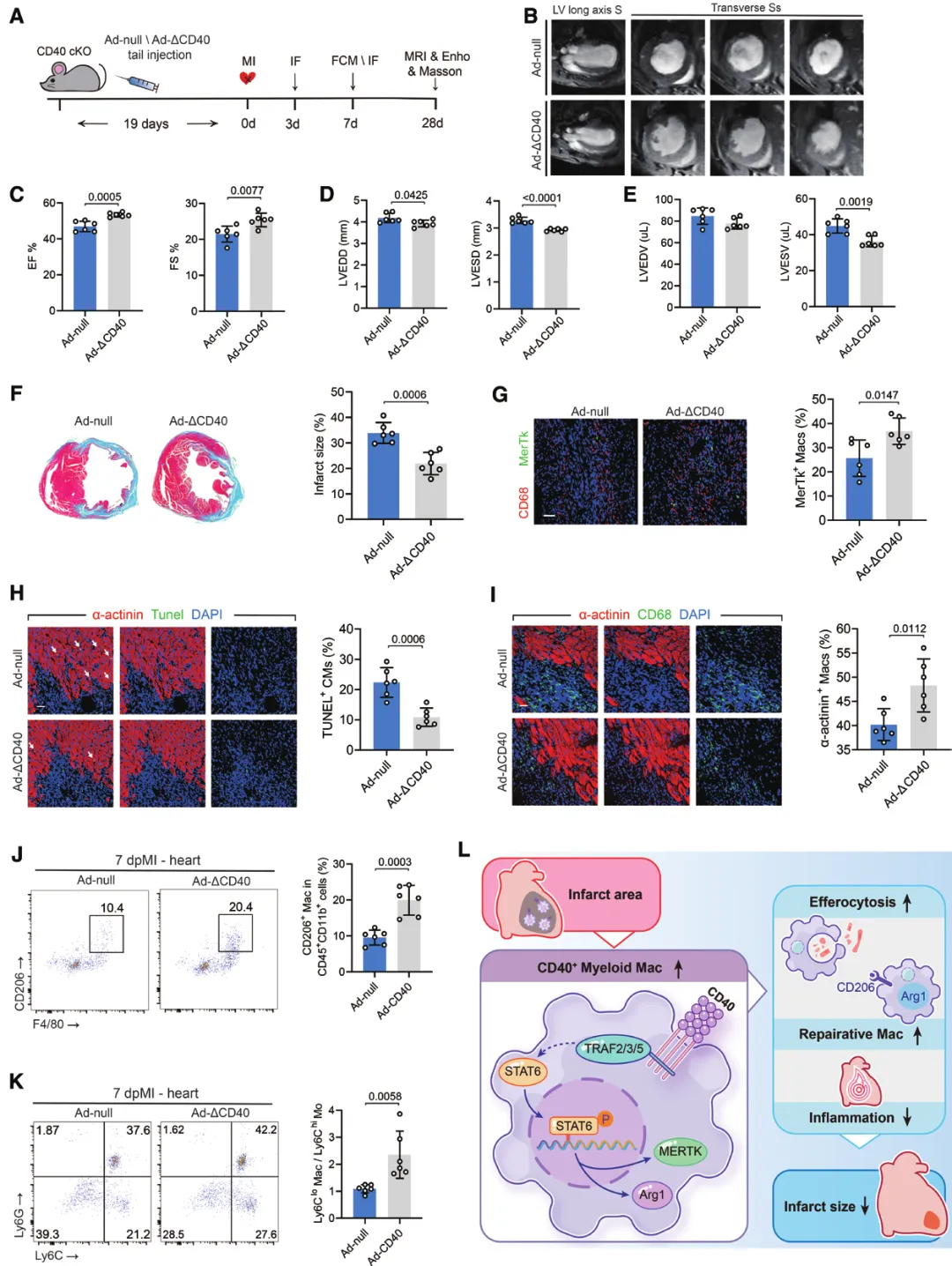
五、临床价值
为动脉粥样硬化、肥胖相关代谢疾病等其他慢性炎症疾病的免疫治疗提供参考(既往研究已发现该通路在代谢调控中也有重要作用)。
该研究改写了“CD40仅作为促炎分子”的传统认知,明确了CD40-TRAF2/3/5-STAT6-MerTk/Arg1是心梗后巨噬细胞胞葬与心脏修复的关键调控轴,为缺血性心脏病提供了精准免疫干预的理论基础。
本文原创仅指编译原创,文献内容与图片版权归原著所有,文献解读仅用于学术分享,如有侵权请与后台联系

 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
