南京工业大学Small研究论文:通过对 U(VI)具有强亲和力的二维阴离子层的有机杂化银锑硫化物实现的快速光催化还原U(VI)
通过对 U(VI)具有强亲和力的二维阴离子层的有机杂化银锑硫化物实现的快速光催化还原U(VI)
第一作者:Longfei Zhai
通讯作者:Wei-Wei Xiong, Bing Zheng
单位:南京工业大学
链接:https://doi.org/10.1002/smll.73757
由于无碳排放和高能量密度,核能被认为是传统化石燃料的有前景的替代品。不幸的是,在核燃料的开采和使用过程中产生了大量的含铀废水。铀的放射性和毒性造成严重的环境污染问题,限制了核能的可持续发展。值得注意的是,U(VI)在含铀废水中可以以迁移能力很强的铀酰离子(UO22+)存在,进一步加深了铀的危害。因此,将高溶解度的六价铀(UO22+)转化为四价铀(UO2)是一种控制水体铀污染的有前景的策略,同时从水中回收UO2可以节约铀资源。光催化还原技术可以利用太阳能将U(VI)还原为U(IV),从而有机会从废水中去除并回收铀。此外,该技术具有能耗低、无污染和可持续性等特点。然而,目前探索高效光催化剂仍然是光催化U(VI)还原技术发展的关键挑战。
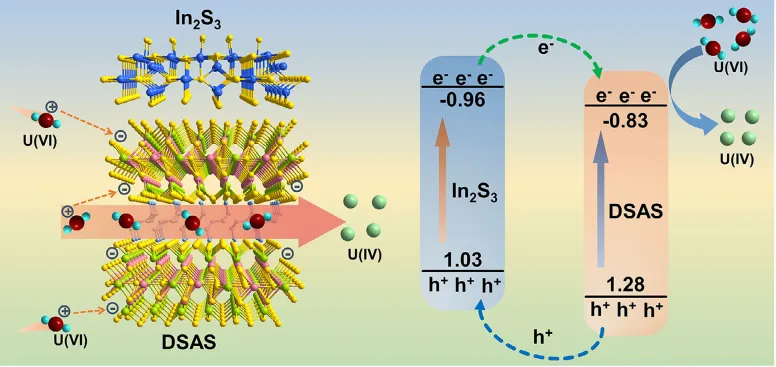
近期,南京工业大学的Wei-Wei Xiong, Bing Zheng等合作在Small发表了题为“Fast Photocatalytic U(VI) Reduction Through an Organic Hybrid Silver Antimony Sulfide Featuring a Two-Dimensional Anionic Layer With Strong Affinity to U(VI)”的研究论文。本文合成了一种有机杂化银锑硫化物[1,4-DABH2][Ag3Sb3S7](1,4-DAB = 1,4-二氨基丁烷),称为DSAS,并用于光催化U(VI)还原。 DSAS具有二维带负电的[Ag3Sb3S7]n2n−阴离子层,可以通过静电相互作用吸引UO2 2+阳离子,从而提高对UO2 2+阳离子的吸附。为了加速DSAS中电子空穴对的分离,通过与In2S3研磨DSAS,制备了一系列DSAS@In2S3-x(x= 15、30、45)复合材料。能带结构和原位辐照XPS证明了DSAS和In2S3之间形成了II型异质结,该异质结在DSAS上积累了光电子以还原U(VI)。因此,DSAS@In2S3-x复合材料表现出出色的光催化U(VI)还原活性。特别是,DSAS@In2S3-30的U(VI)转化率可达312 mg g−1 h−1,高于大多数报道的光催化剂。该研究证明了有机杂化金属硫化物作为光催化剂修复含U(VI)废水的可行性。
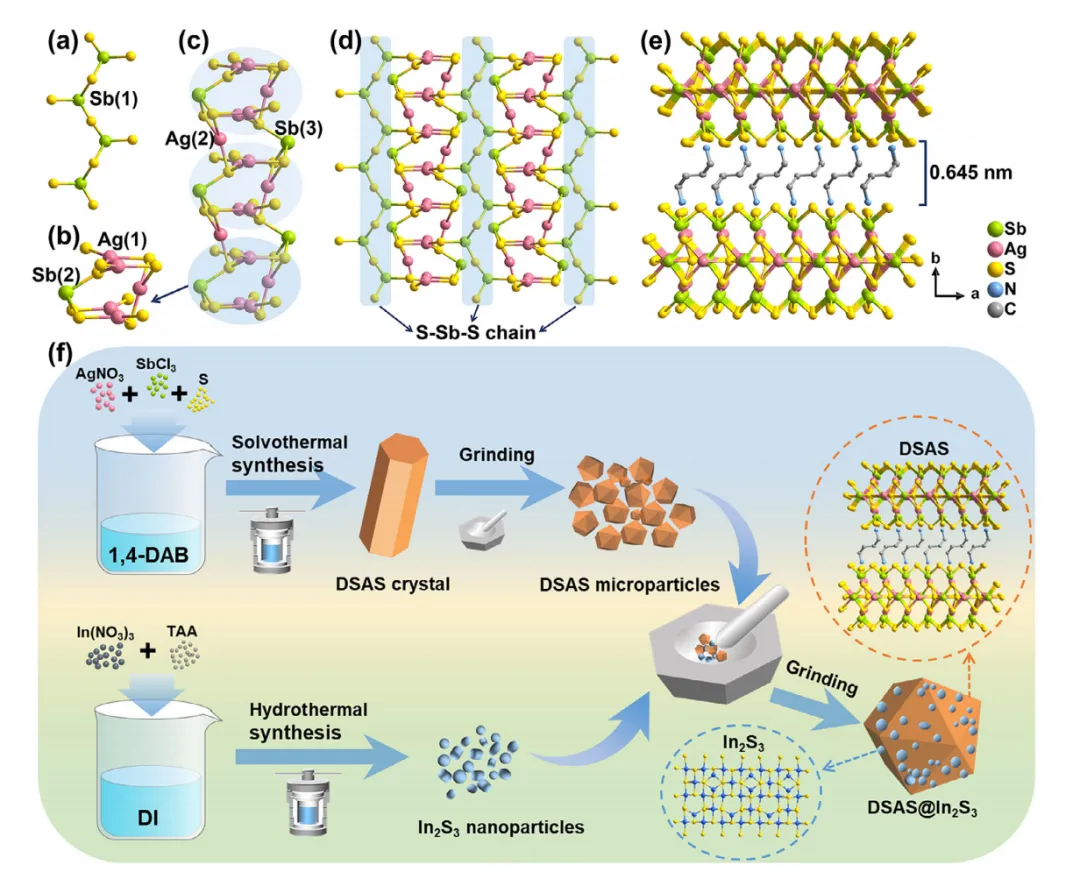
图1 (a) S-Sb-S链。(b) [Ag5SbS10]12−簇。(c) [Ag6Sb2S10]n8n−链。(d) [Ag3Sb3S7]n2n−层。(e) DSAS 的堆叠图。(f) DSAS@In2S3-x的合成过程。
要点一:材料的合成与XRD、SEM表征
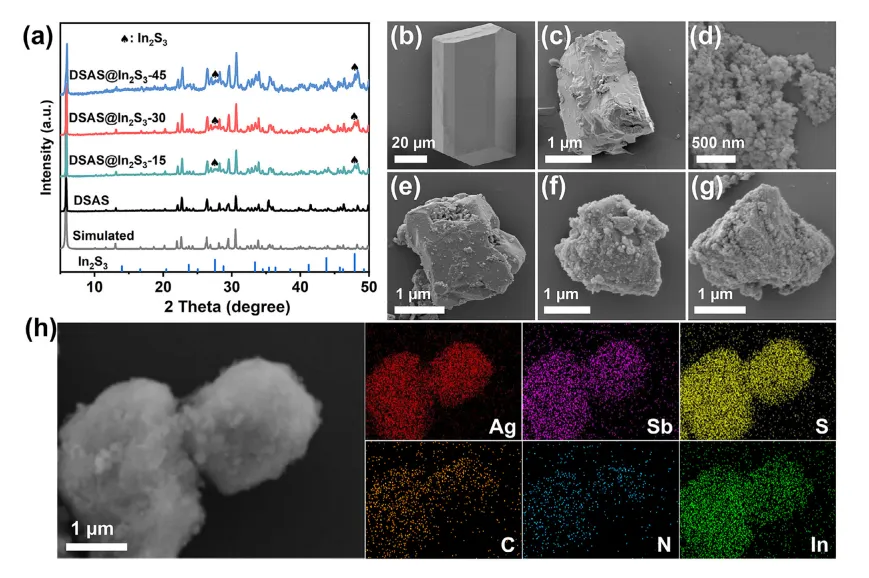
DSAS的棒状晶体是通过溶剂热法合成的,而In2S3纳米粒子是通过水热法制备的。然后,通过湿磨将DSAS晶体转化为微粒。之后,将不同质量比的DSAS微粒和In2S3纳米颗粒在研钵中混合,并加入乙醇研磨30分钟。最后,获得了DSAS@In2S3-x(x = 15、30、45)复合材料,其中x指的是在制备DSAS@In2S3-x时添加的In2S3的质量。图2a中的PXRD图案表明,研磨后DSAS的结构可以保持。此外,DSAS@In2S3-x复合材料的衍射峰同时包含DSAS和In2S3的特征峰,证明DSAS@In2S3-x复合材料的制备成功。
通过SEM表征来记录从DSAS晶体到DSAS@In2S3-x复合材料的形貌变化。通过溶剂热法制备的DSAS晶体是平均直径约为50 µm的微棒(图2b)。研磨后,微棒被粉碎成尺寸约为2 µm的微粒(图2c)。通过水热法制备的In2S3纳米颗粒发生聚集(图2d)。DSAS@In2S3-15复合材料是通过将15毫克In2S3纳米颗粒与90毫克DSAS微粒研磨得到的。DSAS@In2S3-15的 SEM 图像显示,研磨后DSAS微粒保持其尺寸和形状,但一些In2S3纳米颗粒附着在DSAS 微粒的表面(图 2e)。当In2S3纳米粒子与DSAS微粒的质量比增加到30:90时,所制备的复合材料被命名为DSAS@In2S3-30。DSAS@In2S3-30的SEM 分析表明,与DSAS@In2S3-15 相比,更多的In2S3纳米颗粒附着在DSAS 微粒表面(图 2f)。DSAS@In2S3-45由In2S3纳米粒子和DSAS微粒以质量比45:90研磨而成。
图2g显示了DSAS@In2S3-45 的 SEM 图像。显然,聚集的In2S3纳米颗粒形成了厚厚的一层,覆盖在DSAS微粒的表面。如此厚的In2S3层可以抑制DSAS的光吸收。DSAS@In2S3-30的EDX图谱测试表明,银、锑、铟、碳、氮和硫元素均匀分布在微粒上(图2h)。铟元素的均匀分布证实了In2S3纳米颗粒在DSAS表面的均匀附着,进一步证明了DSAS@In2S3-30复合材料的成功合成。
图2. (a) DSAS 和DSAS@In2S3-x的PXRD图谱。 (b) DSAS 块晶体的SEM、DSAS。(c)微粒、(d)In2S3纳米颗粒、(e)DSAS@In2S3-15、(f)DSAS@In2S3-30和(g)DSAS@In2S3-45、(h) DSAS@In2S3-30的EDX映射图像。
要点二:材料的XPS表征
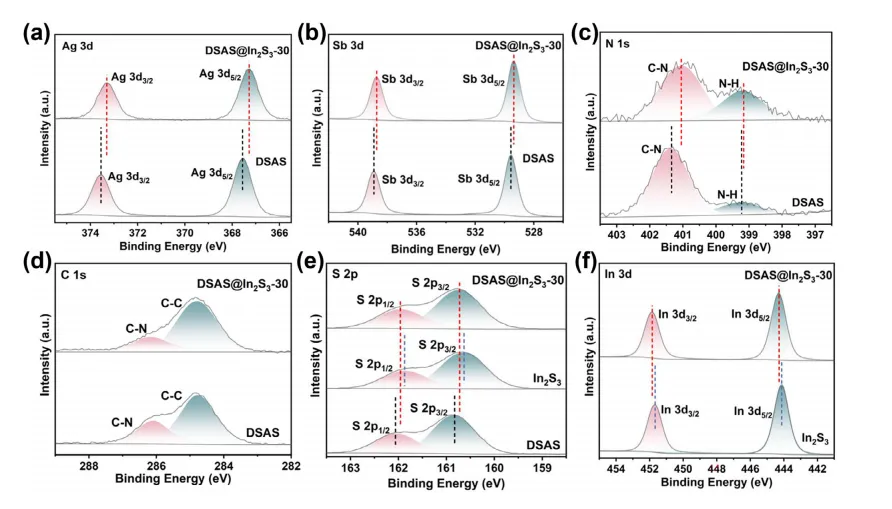
为了进一步证明DSAS和In2S3之间的相互作用,对DSAS、In2S3和DSAS@In2S3-30复合材料进行了XPS测试。XPS调查光谱显示DSAS@In2S3-30中存在银、锑、碳、氮、硫和铟,这进一步证明了In2S3纳米颗粒的成功附着。在 DSAS 的 Ag 3d XPS谱中,Ag 3d3/2和 Ag 3d5/2的两个峰被分配给Ag+(图 3a)。相反,DSAS@In2S3-30光谱中的这两个峰移动到较低的结合能。此外,与DSAS的两个Sb 3d峰和两个N 1s峰相比,DSAS@In2S3-30的Sb 3d峰和N 1s峰也向较低结合能方向移动(图3b,c)。在C 1s XPS谱中,DSAS和DSAS@In2S3-30具有几乎相同的峰位置,对应于C─N和C─C键(图3d)。在S 2p XPS谱中,与DSAS相比,DSAS@In2S3-30的两个峰仍然向较低的结合能移动。然而,与In2S3的两个S 2p峰相比,DSAS@In2S3-30的两个S 2p峰均向更高的结合能方向移动(图3e)。此外,与In2S3的In 3d3/2和In 3d5/2峰相比,DSAS@In2S3-30的In 3d3/2和In 3d5/2峰向更高的结合能移动(图3f)。对于DSAS@In2S3-30,Ag 3d、Sb 3d 和 N 1s XPS 光谱来自DSAS,而 In 3d XPS 光谱来自 In2S3。显然,DSAS的峰转向较低的结合能,而In2S3的峰转向较高的结合能。该结果证实了 DSAS和In2S3之间的相互作用,表明DSAS/In2S3异质结的形成。
图3. DSAS和DSAS@In2S3-30的Ag3d (a)、Sb 3d (b)、N 1s (c) 和 C 1s (d) XPS光谱。 (e) DSAS、In2S3和DSAS@In2S3-30的S 2p XPS 光谱。(f) In2S3和DSAS@In2S3-30的In 3d XPS 光谱。
要点三:光催化性能
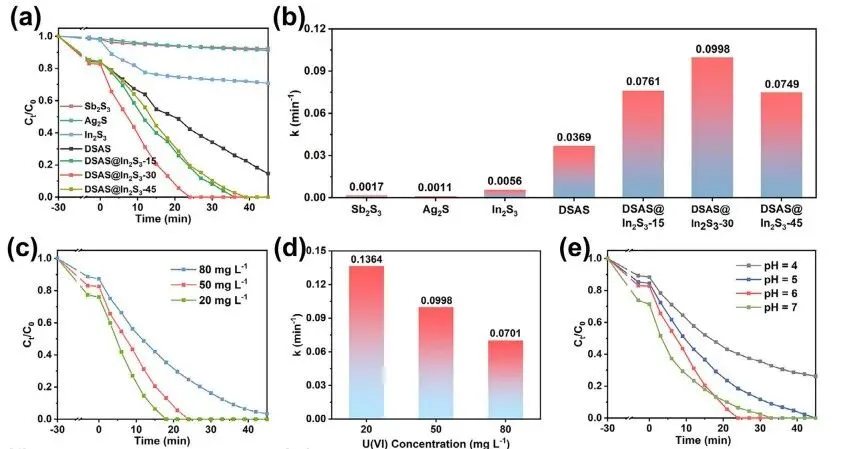
如图5a所示,在30 min的暗反应过程中,无机金属硫化物Sb2S3、Ag2S和In2S3分别可以吸附2.0%、1.5%和2.6%的U(VI),远低于DSAS(16.1%)。 DSAS优异的吸附能力主要归功于其独特的二维阴离子层状结构,这与上述讨论一致。 DSAS晶体在高浓度(200 mg L−1)的U(VI)溶液中吸附后进行了XRD测试。吸附后,对应于DSAS层间晶面的衍射峰向较小角度移动。根据布拉格定律2dsinθ = nλ衍射峰向较小角度的偏移表明DSAS中[Ag3Sb3S7]n2n−阴离子层在U(VI)吸附后层间距离变大。层间距离的增大可能是由于U(VI)吸附过程中UO22+阳离子嵌入层间所致为了验证这一假设,XPS 深度剖析测试进行了对 U(VI) 吸附后的DSAS 块状晶体进行了实验。
同时,图 5b 提供了光催化反应动力学速率的 k 值。显然,DSAS微粒的光催化U(VI)还原速率常数为0.0369 min−1,分别比Sb2S3纳米棒(0.0017 min−1)和Ag2S纳米棒(0.0011 min−1)高21.7和33.5倍。构建DSAS和In2S3异质结后,DSAS@In2S3-x复合材料的光催化性能进一步提高。特别是,DSAS@In2S3-30的光催化U(VI)还原速率常数为0.0998 min−1,分别是DSAS微粒和In2S3纳米粒子的2.7和17.8倍。
如图5c所示,当UO22+浓度为20 mg L−1时,DSAS@In2S3-30在黑暗条件下吸附了24%的UO22+离子,照射18 min后可完全还原U(VI)。随着UO22+离子浓度增加到50 mg L−1,DSAS@In2S3-30对U(VI)的吸附率下降到17.4%,需要照射24 min才能完全还原U(VI)。随着UO22+离子浓度增加到80 mg L−1,DSAS@In2S3-30对U(VI)的吸附率(12.7%)下降,照射45 min内U(VI)减少了96.6%。为了进一步比较不同U(VI) 浓度下的光催化速率,图 5d 中提供了指光催化反应动力学速率的 k 值。当UO22+离子浓度为20,50和80mgL−1时,DSAS@In2S3-30的光催化反应速率常数分别为0.1364、0.0998和0.0701 min−1。 U(VI)浓度的增加需要DSAS@In2S3-30的活性位点参与催化反应,而保持DSAS@In2S3-30用量恒定不可避免地会导致光催化反应速率的下降。考察了不同pH值下DSAs@In2S3-30对铀(VI)的光催化还原性能。当U(VI)溶液的pH值为4、5、6和7时,DSA@In2S3-30照射24分钟后的U(VI)降解率分别为59.7%、81.5%、100%和93.5%(图5e)。
图5. (a)光催化U(VI)还原性能。(b)Sb2S3、Ag2S、In2S3、DSAS和DSAS@In2S3-x的反应速率常数。(c)DSAS@In2S3-30在不同U(VI)浓度下的光催化活性及(d)反应速率常数。(e)DSAS@In2S3-30在不同pH值下的光催化活性。
要点四:光催化机理
基于独特的二维阴离子层,DSAS可以通过静电相互作用吸附更多的UO22+阳离子,从而加速催化反应动力学。同时,较大的层间距离允许UO22+阳离子传输到层间,因此位于层上的额外活性位点可用于U(VI)还原。DSAS与In2S3结合后,形成DSAS与In2S3之间的II型异质结。在可见光照射下,DSAS和In2S3都会产生光生电子和空穴。在DSAS与In2S3形成的II型异质结中,In2S3的CB电位高于DSAS。原位辐照XPS测试证实,来自In2S3CB的光生电子可以自发转移到 DSAS CB。同时,由于DSAS的VB电势高于In2S3的VB电势,来自DSAS的VB的空穴可以转移到In2S3的VB上。因此,光生电子和空穴被有效分离,限制光生载流子的复合。光生电子在DSAS中聚集,还原U(VI);电子与O2反应形成.O2−自由基,该自由基也参与 U(VI) 的还原。因此,在DSAS@In2S3-30光催化U(VI)还原中,光生电子聚集在DSAS中,而UO2 2+ 阳离子由于其带负电的[Ag3Sb3S7]n 2n−层也被DSAS吸引。在这种情况下,更多的光生电子可用于光催化U(VI)还原。由于这些因素,DSAS@In2S3-30复合材料可以表现出出色的光催化性能。
图6:DSAS@In2S3-30还原U(VI) 的光催化机制
综上所述,本文用溶剂热合成了无机杂化银锑硫化物 [1,4-DABH2][Ag3Sb3S7] (DSAS),并用于光催化 U(VI) 还原。 DSAS的特点是二维层状结构[Ag3Sb3S7]n2n-,其中双质子化的1,4-DAB阳离子位于层间。阴离子层[Ag3Sb3S7]n2n-之间的距离达到0.645 nm。如此大的层间距离为UO22+阳离子传输到层间提供了足够的空间,从而为 U(VI) 还原提供了位于层间的额外活性位点。同时带负电的[Ag3Sb3S7]n2n-层可以通过静电相互作用吸引更多的UO22+阳离子,从而提高UO22+阳离子的吸附,从而加速光催化反应动力学。这些特性赋予DSAS高效光催化还原 U(VI) 的潜力。为了进一步提高光催化性能,通过将DSAS微粒与In2S3纳米粒子一起研磨,制备了一系列DSAS@In2S3-x复合材料。通过能带结构测量和原位辐照XPS测试,证实了DSAS和In2S3构建的II型异质结。正如预期的那样,II型异质结的构建提高了光生载流子的分离效率,TPR和PL测试证明了这一点。与DSAS和In2S3相比,所制备的DSAS@In2S3-x表现出优异的U(VI)还原光催化性能。特别是,DSAS@In2S3-30可以在24分钟内完全去除U(VI) (50 mg L−1)。其U(VI)还原率常数分别比DSAS和In2S3高2.7和17.8倍。此外,DSAS@In2S3-30的U(VI)转化率(312mg g−1h−1)远高于大多数报道的光催化剂。本文的研究表明,有机杂化金属硫化物可以作为U(VI)还原的高效光催化剂。
Fast Photocatalytic U(VI) Reduction Through an Organic Hybrid Silver Antimony Sulfide Featuring a Two-Dimensional Anionic Layer With Strong Affinity to U(VI)
https://doi.org/10.1002/smll.73757
整理:库静文
编辑:张湘媛
欢迎关注我们,订阅更多最新消息
点击左下角“阅读原文”即可查看论文原文