胶质母细胞瘤 (GBM) 是侵袭性最强、治疗抵抗最突出的原发性脑肿瘤之一,临床一线方案主要依赖电离辐射 (IR) 与替莫唑胺 (TMZ),但疗效仍受到肿瘤复发和细胞异质性的限制。胶质母细胞瘤干细胞 (GSCs) 具有自我更新、多向分化和强治疗耐受能力,被认为是 GBM 难治的重要细胞基础。与此同时,癌细胞对铁的需求普遍升高,理论上更容易发生铁依赖性脂质过氧化驱动的铁死亡,但 GSCs 虽然摄铁能力增强,却表现出明显的铁死亡抵抗。
 https://www.nature.com/articles/s41556-026-01953-5
https://www.nature.com/articles/s41556-026-01953-5南京医科大学钱旭团队,在 Nature Cell Biology 发表题为 Stress granules restrain ferroptosis by sequestering ferritin 的研究。核心结论是,IR/TMZ 诱导 GSCs 形成 SGs,SGs 通过招募铁蛋白限制铁死亡,从而维持 GSCs 的治疗压力适应。
机制上,SGs 核心蛋白 Ras GTP 酶激活蛋白结合蛋白 1 (G3BP1) 在 IR/TMZ 诱导下发生 333 位甲硫氨酸 (M333) 氧化,进而直接结合铁蛋白轻链 (FTL),并带动铁蛋白重链 (FTH1) 一同进入 SGs。这一过程降低不稳定铁池 (LIP) 中的 Fe2+,并限制核受体共激活因子 4 (NCOA4) 介导的铁蛋白自噬。进一步筛选得到的小分子刺五加苷 C3 (CWJ C3) 能破坏 G3BP1 与 FTL 结合,使 GSCs 重新对 IR/TMZ 诱导的铁死亡敏感。
应激颗粒对应对放化疗时维持胶质母细胞瘤干细胞至关重要
首先,从临床单细胞转录组和 GSCs 模型切入,SGs 核心组分在 GSCs 中更高,IR 或 TMZ 可快速诱导 G3BP1 阳性颗粒。G3BP1/2 双敲除 (dKO) 在常规培养下对干性影响有限,但在治疗压力下,自我更新、神经球形成、增殖和干性标志均明显下降。原位脑瘤模型中,dKO 细胞对 IR/TMZ 更敏感,小鼠生存延长,说明 SGs 是治疗压力下维持 GSCs 的关键缓冲系统。
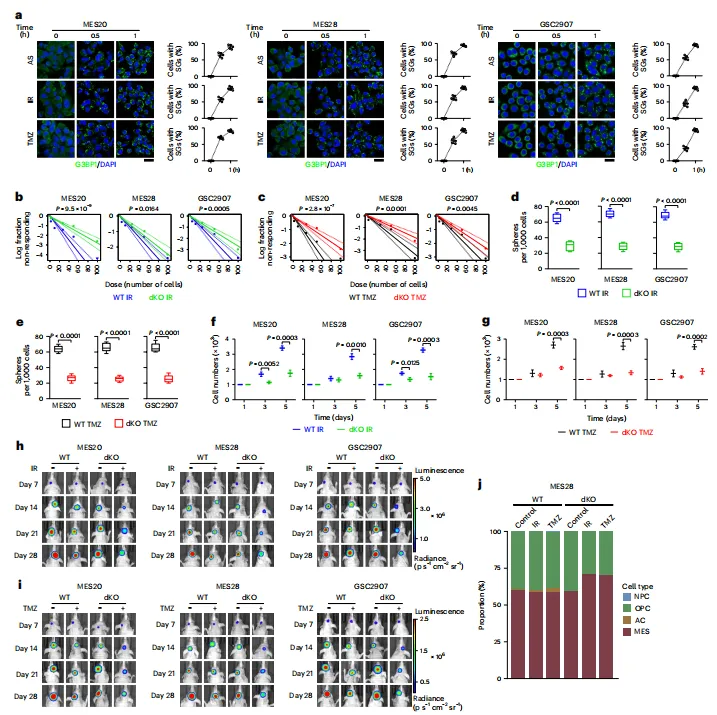 图例:图 a 显示 AS、IR 和 TMZ 诱导 MES20、MES28 与 GSC2907 中 G3BP1 阳性 SGs 的形成;图 b、c 显示 IR 或 TMZ 处理后的体外极限稀释分析;图 d、e 显示每 1,000 个 GSCs 形成的神经球数量;图 f、g 显示细胞增殖变化;图 h、i 显示颅内移植瘤在 IR 或 TMZ 处理后的生物发光信号;图 j 显示 MES28 细胞状态组成变化。
图例:图 a 显示 AS、IR 和 TMZ 诱导 MES20、MES28 与 GSC2907 中 G3BP1 阳性 SGs 的形成;图 b、c 显示 IR 或 TMZ 处理后的体外极限稀释分析;图 d、e 显示每 1,000 个 GSCs 形成的神经球数量;图 f、g 显示细胞增殖变化;图 h、i 显示颅内移植瘤在 IR 或 TMZ 处理后的生物发光信号;图 j 显示 MES28 细胞状态组成变化。应激颗粒限制应激诱导的铁死亡
随后,SGs 蛋白组成被系统解析,IR、TMZ 与亚砷酸钠 (AS) 诱导的 SGs 共享 625 个相关蛋白,富集到细胞死亡通路,铁死亡位列其中。在 G3BP1/2 dKO GSCs 中,IR/TMZ 诱导的死亡可被 Liproxstatin-1 (Lipro-1) 或去铁胺 (DFO) 明显阻断,而凋亡和坏死抑制剂作用有限。线粒体嵴减少、脂质过氧化升高和动物组织中 MDA 增强共同说明,SGs 主要通过压制铁死亡保护 GSCs。
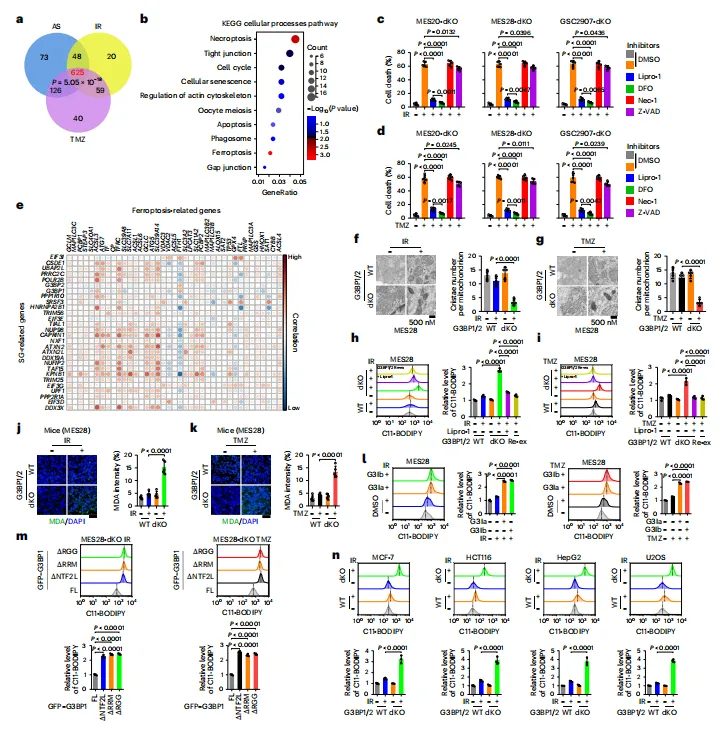 图例:图 a 展示 AS、IR 和 TMZ 诱导的 G3BP1-TurboID 标记蛋白交集;图 b 显示 625 个共有 SGs 相关蛋白的 KEGG 富集;图 c、d 显示不同细胞死亡抑制剂对 dKO GSCs 死亡的影响;图 e 显示 SGs 相关基因与铁死亡相关基因的相关性;图 f、g 显示线粒体超微结构;图 h、i 显示 C11-BODIPY 检测的脂质过氧化;图 j、k 显示动物肿瘤组织 MDA 染色;图 l、m 显示抑制 SGs 形成或表达 G3BP1 突变体后的脂质过氧化;图 n 显示多种肿瘤细胞中的一致现象。
图例:图 a 展示 AS、IR 和 TMZ 诱导的 G3BP1-TurboID 标记蛋白交集;图 b 显示 625 个共有 SGs 相关蛋白的 KEGG 富集;图 c、d 显示不同细胞死亡抑制剂对 dKO GSCs 死亡的影响;图 e 显示 SGs 相关基因与铁死亡相关基因的相关性;图 f、g 显示线粒体超微结构;图 h、i 显示 C11-BODIPY 检测的脂质过氧化;图 j、k 显示动物肿瘤组织 MDA 染色;图 l、m 显示抑制 SGs 形成或表达 G3BP1 突变体后的脂质过氧化;图 n 显示多种肿瘤细胞中的一致现象。应激颗粒通过限制不稳定铁池抑制铁死亡
接着,铁死亡上游的铁稳态被进一步定位。G3BP1/2 缺失没有明显改变谷胱甘肽 (GSH) 或 GPX4、FSP1、DHODH、GCH1 等脂质过氧化防御蛋白,却显著提高 LIP 和 Fe2+。Calcein-AM、u-Ferene 和 FerroOrange 结果一致表明,SGs 缺失使 IR/TMZ 后可反应铁释放增加,尤其是溶酶体 Fe2+ 增高。恢复 G3BP1/2 表达可逆转这一变化,提示 SGs 限制铁死亡的关键入口是压低 LIP 与 Fe2+。
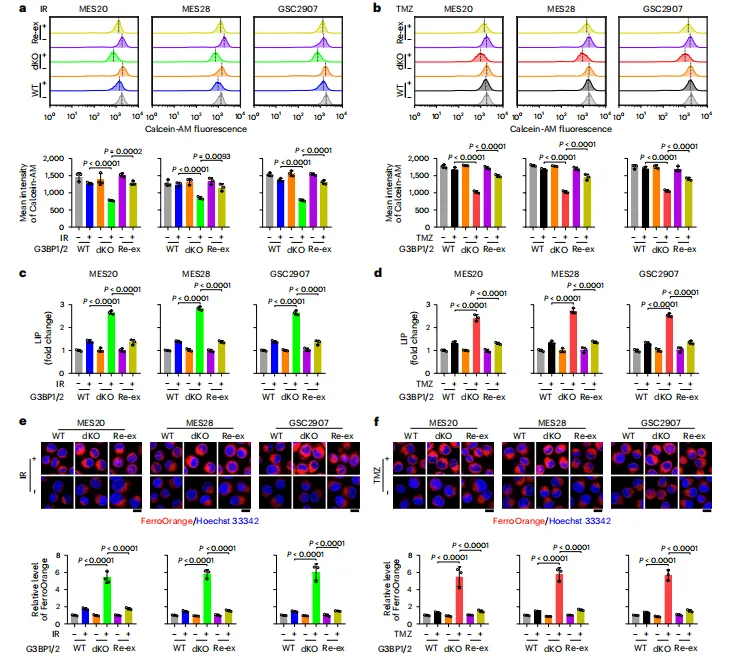 图例:图 a、b 显示 Calcein-AM 流式检测 IR 或 TMZ 后的细胞内不稳定铁;图 c、d 显示 u-Ferene 比色法检测 LIP;图 e、f 显示 FerroOrange 探针检测 Fe2+,并比较 WT、dKO 和 G3BP1/2 重新表达细胞在 IR 或 TMZ 处理后的变化。
图例:图 a、b 显示 Calcein-AM 流式检测 IR 或 TMZ 后的细胞内不稳定铁;图 c、d 显示 u-Ferene 比色法检测 LIP;图 e、f 显示 FerroOrange 探针检测 Fe2+,并比较 WT、dKO 和 G3BP1/2 重新表达细胞在 IR 或 TMZ 处理后的变化。G3BP1 直接与 FTL 相互作用
随后,SGs 如何限制 LIP 被聚焦到铁蛋白。蛋白组结果提示 FTL 与 FTH1 是与 G3BP1 同时富集的重要铁相关蛋白。IR/TMZ 处理后,GFP-G3BP1-TurboID 对 FTL 和 FTH1 的邻近标记增强,内源性免疫沉淀确认 G3BP1 与 FTL 结合。删除 FTL 会阻断 G3BP1 与 FTH1 的连接,而删除 FTH1 不影响 G3BP1 与 FTL 结合,说明 G3BP1 直接识别 FTL,FTL 是连接 G3BP1 与完整铁蛋白复合体的桥梁。
 图例:图 a、b 显示 IR 或 TMZ 后链霉亲和素 pull-down 检测 FTL、FTH1 与 G3BP1 邻近标记增强;图 c、d 显示 G3BP1 与 FTL 的内源性互作;图 e、f 比较 FTL 或 FTH1 缺失后 G3BP1 与铁蛋白组分的结合;图 g 显示 G3BP2 与 FTL 的应激依赖性结合依赖 G3BP1。
图例:图 a、b 显示 IR 或 TMZ 后链霉亲和素 pull-down 检测 FTL、FTH1 与 G3BP1 邻近标记增强;图 c、d 显示 G3BP1 与 FTL 的内源性互作;图 e、f 比较 FTL 或 FTH1 缺失后 G3BP1 与铁蛋白组分的结合;图 g 显示 G3BP2 与 FTL 的应激依赖性结合依赖 G3BP1。G3BP1 将铁蛋白募集至应激颗粒
在确定直接互作后,铁蛋白的空间去向成为关键。IR/TMZ 诱导 FTL 与 FTH1 进入 G3BP1 阳性 SGs,且 FTL 进入 SGs 的动态与 G3BP1 同步。G3BP1 缺失比 G3BP2 缺失更显著削弱 FTL 招募,并造成更高 LIP 与脂质过氧化。FTL 缺失还会阻断 FTH1 和 NCOA4 进入 SGs,而 SGs 缺失增强 FTH1 与 LC3 阳性结构共定位,说明 SGs 通过 G3BP1-FTL 轴限制 NCOA4 介导的铁蛋白自噬。
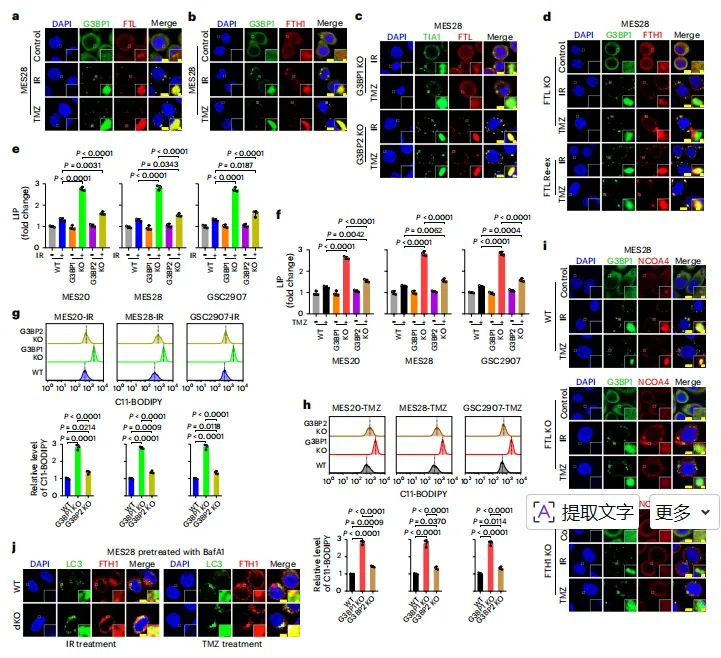 图例:图 a、b 显示 IR 或 TMZ 后 FTL、FTH1 与 G3BP1 阳性 SGs 的共定位;图 c 显示 G3BP1 或 G3BP2 缺失对 FTL 募集的影响;图 d 显示 FTL 缺失及重新表达对 FTH1 进入 SGs 的影响;图 e、f 显示 G3BP1 或 G3BP2 缺失后的 LIP;图 g、h 显示对应的脂质过氧化;图 i 显示 NCOA4 进入 SGs 对 FTL 和 FTH1 的依赖;图 j 显示 SGs 缺失后 FTH1 与 LC3 阳性铁蛋白自噬结构增加。
图例:图 a、b 显示 IR 或 TMZ 后 FTL、FTH1 与 G3BP1 阳性 SGs 的共定位;图 c 显示 G3BP1 或 G3BP2 缺失对 FTL 募集的影响;图 d 显示 FTL 缺失及重新表达对 FTH1 进入 SGs 的影响;图 e、f 显示 G3BP1 或 G3BP2 缺失后的 LIP;图 g、h 显示对应的脂质过氧化;图 i 显示 NCOA4 进入 SGs 对 FTL 和 FTH1 的依赖;图 j 显示 SGs 缺失后 FTH1 与 LC3 阳性铁蛋白自噬结构增加。G3BP1 的 M333 氧化促进其与 FTL 相互作用
机制继续推进到 G3BP1 的氧化修饰。结构域截短与分子对接显示,G3BP1 的 PxxP 区域参与 FTL 识别,其中 M333 对结合最关键。IR/TMZ 增加活性氧 (ROS),而甲硫氨酸还原酶 A (MsrA) 会削弱 G3BP1-FTL 互作并增强脂质过氧化。Ox4 探针、氧化还原激活化学标记 (ReACT) 和质谱共同支持 G3BP1 M333 被氧化。模拟氧化的 M333Q 促进 FTL 募集,M333A 则失效,证明M333 氧化是 SGs 限制铁死亡的分子开关。
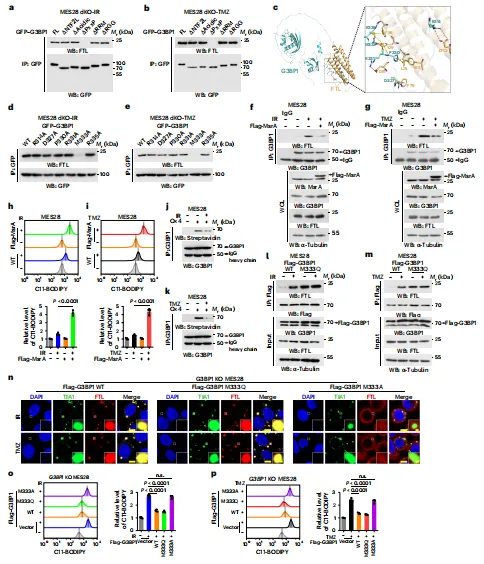 图例:图 a、b 显示 G3BP1 不同结构域缺失体与 FTL 的结合;图 c 显示 G3BP1 与 FTL 的对接模型及关键残基;图 d、e 显示多个点突变对互作的影响;图 f、g 显示 MsrA 对 G3BP1-FTL 结合的抑制;图 h、i 显示 MsrA 过表达后的脂质过氧化;图 j、k 显示 Ox4 检测 G3BP1 甲硫氨酸氧化;图 l、m 显示 M333Q 对互作的促进;图 n 显示 WT、M333Q 和 M333A 对 FTL 募集的影响;图 o、p 显示对应的铁死亡水平。
图例:图 a、b 显示 G3BP1 不同结构域缺失体与 FTL 的结合;图 c 显示 G3BP1 与 FTL 的对接模型及关键残基;图 d、e 显示多个点突变对互作的影响;图 f、g 显示 MsrA 对 G3BP1-FTL 结合的抑制;图 h、i 显示 MsrA 过表达后的脂质过氧化;图 j、k 显示 Ox4 检测 G3BP1 甲硫氨酸氧化;图 l、m 显示 M333Q 对互作的促进;图 n 显示 WT、M333Q 和 M333A 对 FTL 募集的影响;图 o、p 显示对应的铁死亡水平。CWJ C3 破坏 G3BP1 与 FTL 的相互作用
在机制基础上,研究进一步寻找可干预分子。围绕 FTL 的 G3BP1 结合界面,对 56,883 个中枢神经系统 (CNS) 可穿透或靶向化合物进行虚拟筛选,CWJ C3 显示较高 FTL 亲和力。表面等离子共振 (SPR) 与微量热泳动 (MST) 结果表明,CWJ C3 可结合 FTL 并削弱 G3BP1-FTL 结合。在细胞中,CWJ C3 减少 FTL 进入 SGs,并增强 IR/TMZ 后 Fe2+ 积累,说明它能够解除 SGs 对铁池的限制,把保护性隔离转化为促铁死亡信号。
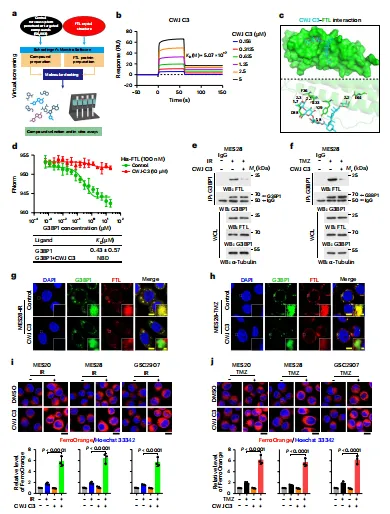 图例:图 a 显示以 FTL 为靶点的 CNS 可穿透化合物虚拟筛选流程;图 b 显示 SPR 测定 CWJ C3 与 FTL 的结合;图 c 显示 FTL-CWJ C3 对接模型;图 d 显示 MST 检测 CWJ C3 对 G3BP1-FTL 结合的影响;图 e、f 显示细胞中 CWJ C3 对 IR 或 TMZ 诱导互作的抑制;图 g、h 显示 FTL 进入 SGs 减少;图 i、j 显示 FerroOrange 检测到 Fe2+ 升高。
图例:图 a 显示以 FTL 为靶点的 CNS 可穿透化合物虚拟筛选流程;图 b 显示 SPR 测定 CWJ C3 与 FTL 的结合;图 c 显示 FTL-CWJ C3 对接模型;图 d 显示 MST 检测 CWJ C3 对 G3BP1-FTL 结合的影响;图 e、f 显示细胞中 CWJ C3 对 IR 或 TMZ 诱导互作的抑制;图 g、h 显示 FTL 进入 SGs 减少;图 i、j 显示 FerroOrange 检测到 Fe2+ 升高。CWJ C3 通过诱导铁死亡提高 GSCs 对放化疗的敏感性
最后,药理干预的功能意义被放回 GSCs 与动物模型中验证。CWJ C3 单独作用毒性有限,但与 IR/TMZ 联用显著增加细胞死亡和脂质过氧化,并削弱极限稀释实验和神经球实验反映的自我更新能力。在 G3BP1 M333A 背景下,CWJ C3 不再进一步增强铁死亡,说明其效应依赖 G3BP1-FTL 轴。颅内移植瘤中,CWJ C3 联合 IR 或 TMZ 抑制肿瘤生长、延长生存并提高 MDA 信号,且主要器官未见明显毒性,提示该轴具备治疗转化潜力。
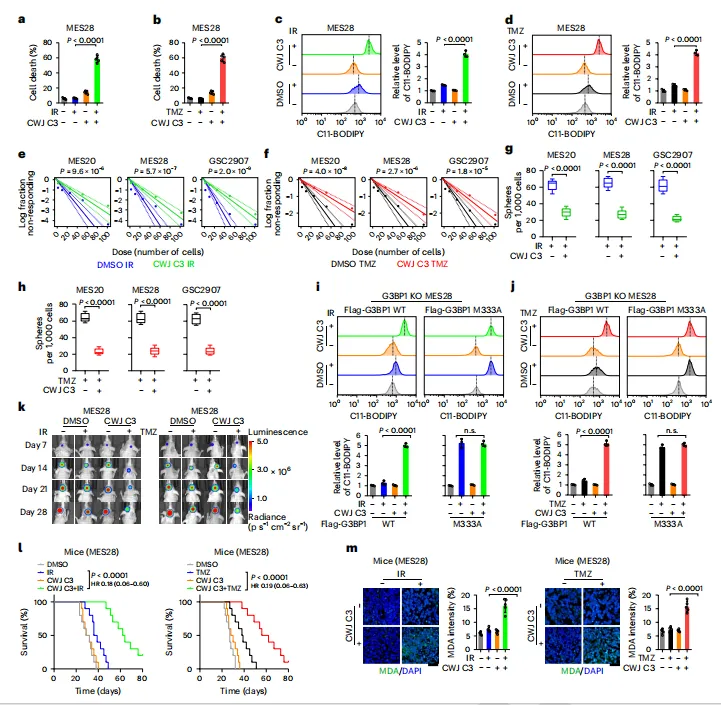 图例:图 a、b 显示 CWJ C3 联合 IR 或 TMZ 后的细胞死亡;图 c、d 显示 C11-BODIPY 检测的脂质过氧化;图 e、f 显示体外极限稀释实验;图 g、h 显示神经球形成能力;图 i、j 显示 G3BP1 WT 或 M333A 重构细胞中 CWJ C3 对铁死亡的影响;图 k 显示颅内移植瘤生物发光成像;图 l 显示小鼠生存曲线;图 m 显示肿瘤组织 MDA 免疫组化结果。
图例:图 a、b 显示 CWJ C3 联合 IR 或 TMZ 后的细胞死亡;图 c、d 显示 C11-BODIPY 检测的脂质过氧化;图 e、f 显示体外极限稀释实验;图 g、h 显示神经球形成能力;图 i、j 显示 G3BP1 WT 或 M333A 重构细胞中 CWJ C3 对铁死亡的影响;图 k 显示颅内移植瘤生物发光成像;图 l 显示小鼠生存曲线;图 m 显示肿瘤组织 MDA 免疫组化结果。总结
研究揭示了一条 SGs 限制铁死亡的新机制:IR/TMZ 使 G3BP1 的 M333 位点发生氧化,增强其与 FTL 的结合,进而把铁蛋白及相关铁代谢节点收纳进 SGs,减少 LIP 中 Fe2+ 的可用性,并抑制 NCOA4 介导的铁蛋白自噬。这一机制解释了 GSCs 在高铁需求与铁死亡风险之间如何取得暂时平衡,也说明 SGs 并非单纯的应激标志物,而是治疗压力下重塑铁代谢和细胞命运的功能平台。CWJ C3 对 G3BP1-FTL 互作的药理破坏,为 GBM 以及可能的其他肿瘤提供了一个可探索的放化疗增敏方向。

 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
